Врожденные миопатии
Эти редко встречающиеся заболевания отличаются от мышечных дистрофий наличием специфических гистобиохимических и структурных дефектов мышечной ткани. Заболевание характеризуется непрогрессирующим течением, что, однако, не является правилом. В типичных случаях у младенцев отмечают гипотонию и задержку двигательного развития. Часто наблюдают «грудь сапожника», кифосколиоз, неправильное положение головки тазовой кости и вогнутую стопу (pes cavus). Своевременная диагностика очень важна, так как долгосрочный прогноз и лечение отличаются от таковых при мышечных дистрофиях.
Различают четыре главные формы врожденной миопатии: центрально-стволовую, немалиновую (стержневая) миопатию, миотубулярную (центронуклеарная) миопатию и врожденную диспропорцию волокнистого типа.
Центрально-стволовая болезнь. Это первая из описанных врожденных миопатий, идентифицированная Shy и Magee в 1956 г. Заболевание наследуется по аутосомно-доминантному типу, однако бывают и спорадические случаи. В младенческом возрасте характерными являются гипотония и задержка двигательного развития, однако заболевание может привлечь к себе внимание и лишь во взрослом возрасте, когда появляется мышечная слабость или те или иные изменения скелета. Больные небольшого роста с хрупкой фигурой; аномалии скелета характеризуются врожденной дислокацией бедер, сколиозом, вогнутой стопой (pes cavus) и «грудью сапожника». Слабость мышц лица и конечностей (особенно конечностей) выражена не очень резко. При исследовании биоптата мышцы обнаруживают мышечные волокна с множественными или единичными прерывистыми (дискретными) зонами (центральные массы некротизированной ткани), лишенные окислительных ферментов. Другие лабораторные тесты менее информативны диагностически, так как активность сывороточной КК и данные ЭМГ могут быть нормальными. Больные с этой патологией предрасположены к развитию злокачественной гипертермии (см. гл. 8).
Немалиновая миопатия. Немалиновая миопатия, называемая также врожденной непрогрессирующей нитеобразной миопатией, была описана Shy и сотр. в 1963 г. Наследуется обычно по аутосомно-доминантному типу, но наследование может быть тоже рецессивным или спорадическим. В младенческом возрасте часто появляется гипотония, смерть может наступить вследствие дыхательной недостаточности. Скелетные аномалии могут быть выражены очень резко. Это резко удлиненное лицо; высокое небо, плохо развитая мускулатура, кифосколиоз, «грудь сапожника», мышечная слабость может распространяться на лицо, мягкое небо, конечности. Прогноз заболевания весьма вариабелен: иногда болезнь не прогрессирует, а если прогрессирует, то вынуждает больных пользоваться сидячей каталкой или же приводит к дыхательной недостаточности. При гистологическом исследовании в мышцах обнаруживают пучки нитеподобных или палочкоподобных немалиновых телец, от чего и произошло название данного заболевания. «Палочки» являются дериватами Z-пучковой субстанции, их обычно находят в мышечных волокнах I типа. а в пораженных мышцах, как правило, преобладают мышечные волокна именно этого типа. Активность сывороточной КК может быть нормальной или слегка повышенной. При ЭМГ обычно выявляют миопатию.
Миотубулярная миопатия. Заболевание это было описано Spiro. Shy и Gonatas в 1963 г.
Гистологические изменения при миотубулярной миопатии напоминают эмбриональную стадию развития мышечных «трубочек» в процессе формирования мышечного волокна. Некоторые авторы предпочитают называть заболевание центронуклеарной миопатией, полагая, что мышечные волокна при этом не имеют эмбрионального характера. Заболевание обычно возникает спорадически, но наследование может быть аутосомно-доминантным, рецессивным или рецессивным сцепленным с Х-хромосомой. В младенческом возрасте отмечаются гипотония и мышечная слабость, что может служить причиной смерти. Проявляясь в более старшем возрасте, болезнь может напоминать немалиновую миопатию. У больных при этом бывают узкое, вытянутое лицо, вогнутая стопа, сколиоз. Общая мышечная масса обычно уменьшена. а степень слабости проксимальной и дистальной мускулатуры варьирует. От других врожденных миопатий эта отличается присутствием наружной офтальмоплегии. Течение болезни может быть как прогрессирующим, так и непрогрессирующим. Активность сывороточной КК нормальная или слегка повышена. ЭМГ патологически изменена: потенциалы моторных единиц уменьшены и избыточно «укомплектованы». В биоптате мышцы обнаруживают мышечные волокна с рядами центрально расположенных ядер, часто окруженных чистой перинуклеарной зоной. В большей степени поражаются мышечные волокна I типа, они могут подвергаться атрофии.
Врожденная диспропорция в соотношении типов мышечных волокон. К клиническим проявлениям этого заболевания относят гипотонию, мышечную слабость, замедленное физическое развитие, а также аномалии скелета, как и при других врожденных миопатиях.
Диагноз основан на результатах мышечной биопсии: в биоптате отмечают увеличение числа небольших мышечных волокон I типа и нормальные или гипертрофированные мышечные волокна II типа. Патогенез заболевания малопонятный. Прогноз обычно благоприятный, у многих бальных с возрастом состояние улучшается, хотя двигательные нарушения той или иной степени все же остаются. У отдельных больных мышечная слабость прогрессирует.
Болезни, обусловленные нарушением энергетического метаболизма в мышцах
В скелетной мускулатуре обычно утилизируются два главных источника энергии — жирные кислоты и глюкоза. Следовательно, нарушение утилизации глюкозы или жиров может сопровождаться четкими клиническими проявлениями со стороны мышечной системы. Наиболее тяжелым проявлением данной патологии является острый мышечный болевой синдром, который может привести к тяжелому рабдомиолизу и миоглобинурии. Следует упомянуть также прогрессирующую мышечную слабость, симулирующую мышечную дистрофию. Объяснений существования этих двух различных клинических синдромов нет.
Гликогеноз (болезнь накопления гликогена) и гликолитические дефекты. Существует четыре вида нарушений обмена гликогена (типы II, III, IV и V) и четыре вида расстройства гликолиза (типы VII, IX, Х и XI), которые проявляются существенными нарушениями со стороны скелетной мускулатуры (см. также гл. 313).
Недостаточность кислой мальтазы (тип II гликогеноза). Кислая мальтаза — это лизосомный энзим из группы кислых гидролаз, обладающий -1,4 и -1,6 глюкозидазной активностью: расщепляет гликоген до глюкозы. В то же время роль этого фермента в обмене углеводов определена недостаточно четко. Существует три клинические формы недостаточности кислой мальтазы, каждая из которых наследуется аутосомно-рецессивно. Биохимическая основа различных клинических проявлений этой ферментной недостаточности неясна.
В младенческом возрасте недостаточность кислой мальтазы проявляется как общий гликогеноз. При рождении патологии не находят, но уже вскоре обнаруживают резкую мышечную слабость, кардиомегалию, гепатомегалию и заметное увеличение размеров языка. Накопление гликогена в моторных нейронах спинного мозга, а также в стволе мозга усугубляет мышечную слабость. Такие младенцы обычно умирают в течение первого года жизни.
У детей и взрослых это заболевание проявляется как мышечная дистрофия. Детские формы заболевания характеризуются замедленным развитием ребенка, слабостью проксимальных мышц конечностей, увеличением размеров икроножных мышц. Заболевание может прогрессировать с развитием дыхательной недостаточности; смерть обычно наступает в конце 2-го десятилетия жизни. Может иметь место поражение сердца, однако гепатомегалия и макроглоссия встречаются редко.
Заболевание у взрослых лиц начинается в 3—4-м десятилетиях жизни и ошибочно может быть диагностировано как конечностно-поясная дистрофия или полимиозит. Начальным проявлением заболевания служит дыхательная недостаточность, обусловленная слабостью диафрагмы. Печень, сердце и язык обычно не поражаются. Предположение о диагнозе возникает после исследования мышечного биоптата, в котором обнаруживают вакуоли, содержащие гликоген, и кислую фосфатазу. При электронной микроскопии видно, что гликоген как связан с мембранами, так и свободно располагается в тканях. Окончательный диагноз устанавливают при биохимическом исследовании пораженной мышцы. Активность кислой мальтазы в моче снижена. Уровень сывороточной активности КК может превышать норму в 10 раз. При ЭМГ можно дифференцировать мальтазную недостаточность от мышечной дистрофии по высокочастотным миотоническим разрядам, сопровождающим непродолжительные потенциалы моторных единиц, на фоне фибрилляций и положительных остроконечных потенциалов.
Недостаточность фермента, тормозящего ветвление молекулы гликогена (гликогеноз III типа). Эта довольно легко протекающая детская болезнь проявляется гепатомегалией, замедлением роста и гипогликемией; нерезко выраженную мышечную слабость наблюдают редко. После пубертатного периода выраженность этих симптомов обычно уменьшается или они исчезают совсем, так что мышечная слабость и некоторое уменьшение мышечной массы могут быть связаны просто с уменьшением физической нагрузки из-за плохой к ней толерантности. Предположение о возможном диагнозе возникает тогда, когда после выполнения больным специального упражнения для мышц предплечья в крови не повышается содержание молочной кислоты. Сывороточная активность КК обычно повышена. При ЭМГ выявляют изменения, характерные для миопатии, а также признаки повышенной раздражимости мембран миотоническими импульсами. В биоптате мышцы обнаруживают вакуоли с повышенным содержанием гликогена. Для подтверждения диагноза требуется биохимическое исследование мышцы.
Недостаточность гликоген-ветвящего фермента (гликогеноз IV типа). Недостаточность данного фермента — это очень тяжелая, фатальная патология младенческого возраста, при которой нарушения со стороны скелетной мускулатуры отходят на второй план по сравнению с развитием хронической печеночной недостаточности. Однако мышечная гипотония и атрофия мышц могут навести на мысль о первично мышечном заболевании или о спинально-мышечной атрофии.
Недостаточность мышечной фосфорилазы (гликогеноз V типа). Плохая переносимость физической нагрузки является характерным симптомом недостаточности мышечной фосфорилазы, впервые описанной в 1951 г. Мак-Ардлем (McArdle). Заболевание наследуется аутосомно-рецессивно; мужчины болеют чаще, чем женщины. После пубертатного возраста у больных возникают болезненные мышечные судороги и быстрая утомляемость мышц после интенсивной физической нагрузки — бега, поднятия тяжестей. В литературе описаны варианты заболевания, начинающегося как в младенческом возрасте, так и позднее. Многие больные сообщают о феномене «второго дыхания», наступающего после кратковременного отдыха или после замедления темпа физической нагрузки, что позволяет им в течение долгих лет сохранять двигательную активность. Физическое переутомление у таких больных ведет к развитию рабдомиолиза, миоглобинурии и почечной недостаточности. Постоянная мышечная слабость и прогрессирующая атрофия мышц отмечаются редко, так что при физическом обследовании их в периоды между обострениями болезни патологии обычно не выявляют. Другие органы при данном заболевании не поражаются.
Активность сывороточной КК подвержена значительным колебаниям и может быть повышена даже в бессимптомные периоды. Тест с нагрузкой на мышцы предплечья не сопровождается повышением в крови содержания молочной кислоты. Данные ЭМГ бывают нормальными, если только ее не осуществляют непосредственно после эпизода рабдомиолиза. В биоптате мышцы выявляют «пузырьки», содержащие гликоген под сарколеммой. Наличие недостаточности мышечной фосфорилазы может быть установлено с помощью гистохимической окраски гистологического препарата или при биохимическом исследовании мышечной ткани. Больные могут оставаться в течение жизни достаточно активными при условии воздержания от тех или иных физических перегрузок. Заместительная диетотерапия глюкозой или фруктозой обычно не сопровождается ослаблением симптоматики заболевания.
Недостаточность фосфофруктокиназы (гликогеноз VII типа). Это заболевание напоминает недостаточность мышечной фосфорилазы и также наследуется по аутосомно-рецессивному типу; среди заболевших преобладают лица мужского пола. Те же, что и при фосфорилазной недостаточности, провоцирующие моменты и данные лабораторных исследований. Выявляют этот вид ферментной недостаточности при гистохимической окраске препарата мышцы на фосфофруктокиназу (ФФрК). Для достоверного диагноза необходимо биохимическое исследование мышечных ферментов. У некоторых больных с недостаточностью указанного фермента возможны нерезко выраженный гемолиз, увеличение числа ретикулоцитов в периферической крови, а также повышение содержания билирубина в крови, так как дефицит ФФрК имеет место при этом не только в мышцах, но и в эритроцитах.
Синдромы, связанные с недостаточностью нового гликолитического фермента. Начиная с 1981 г. была идентифицирована недостаточность еще трех гликолитических ферментов: фосфоглицераткиназы (ФГлК) (тип IX), фосфоглицератмутазы (ФГлМ) (тип X) и лактатдегидрогеназы (ЛДГ) (тип XI). Клиническая картина всех этих трех типов ферментной недостаточности идентична. В раннем детстве или в подростковом возрасте после физических перенапряжений у больных возникают эпизоды миоглобинурии и миалгии. Думается, что все эти ферментные дефекты наследуются по аутосомно-рецессивному типу. Активность сывороточной КК может быть повышена как во время обострений заболеваний,
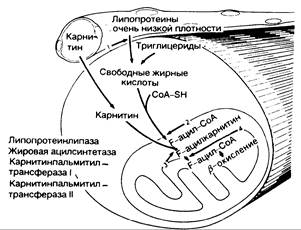
Рис. 357-1. Метаболизм липидов.
Свободные жирные кислоты как энергетическое средство образуются из триглицеридов, накопленных в мышце, и из циркулирующих липопротеинов очень низкой плотности, которые расщепляются под влиянием эндотелиальной липопротеинлипазы (1) в капиллярах. Карнитин, эссенциальный субстрат для метаболизма липидов, образуется в печени и транспортируется к мышце. В мышце свободные жирные кислоты соединяются с коэнзимом A (CoA-SH) под влиянием жирной ацилсинтетазы (2), обнаруживаемой в наружной митохондриальной мембране, в результате чего образуется жировой ацилкоэнзим А (F-ацил-СоА). Транспорт через внутреннюю митохондриальную мембрану требует переноса карнитина с помощью карнитинпальмитинтрансфсразы I (КПТ-1), связанной с наружной поверхностью внутренней митохондриальной мембраны (3). Внутри митохондрии жировой ацилкарнитин (Р-ацилкарнитин) синтезируется с помощью КПТ-П (4), которая связана с внутренней поверхностью внутренней митохондральной мембраны. При этом жировой ацилкоэнзим А подвергается -окислению.
так и между обострениями. При недостаточности ФГлМ и ЛДГ повышение содержания молочной кислоты в крови после нагрузки на мышцы предплечья обычно бывает ниже, чем в норме. При недостаточности ФГлК совсем не повышается содержание в крови лактата после нагрузки. И вообще, эта форма ферментной недостаточности по клиническим проявлениям очень напоминает недостаточность мышечной фосфорилазы и фосфофруктокиназы. Гистологическое исследование мышц при указанных формах энзимной недостаточности обычно неинформативно, отмечается лишь некоторое увеличение содержания в мышцах гликогена. Для достоверной диагностики необходимо биохимическое исследование мышцы.
Нарушения метаболизма липидов. Липиды — это важный энергетический субстрат, особенно во время покоя мышцы и при длительных, но нерезких физических нагрузках (рис. 357-1).
Недостаточность карнитина. Различают миопатическую и системную (генерализованная) формы карнитиновой недостаточности.
Миопатическая карнитиновая недостаточность обычно протекает с генерализованной мышечной слабостью, которая, как правило, начинается в детстве. Клинические проявления этой болезни частично напоминают мышечную дистрофию, а частично — полимиозит. Большинство случаев спорадические; полагают, что болезнь может наследоваться по аутосомно-рецессивному типу. Иногда возникает кардиомиопатия. Активность сывороточной КК слегка повышена; на ЭМГ — признаки миопатии. В биоптате мышцы обнаруживают резко выраженное накопление липидов. Содержание карнитина в сыворотке крови нормальное. Считается, что при этом заболевании нарушается транспорт карнитина в мышцы, поэтому содержание его в мышцах столь низкое. Некоторые больные положительно отвечают на пероральную заместительную терапию карнитином, во всяком случае она должна быть испробована во всех случаях. Другие бальные по неизвестным причинам положительно отвечали на лечение преднизолоном. У некоторых больных лечебный эффект оказала замена в их диете триглицеридов со средней цепью на триглицериды с длинной цепью. Отдельные больные хорошо отвечают на лечение рибофлавином.
Системная карнитиновая недостаточность — это аутосомно-рецессивное заболевание младенческого и раннего детского возраста. Оно характеризуется прогрессирующей мышечной слабостью и эпизодами печеночной энцефалопатии с тошнотой, рвотой, затемнением сознания, комой и ранней смертью. Низкое содержание карнитина в сыворотке крови отличает эту форму от миопатической карнитиновой недостаточности. Не установлено никакой причины, которая могла бы вызывать или объясняла низкое содержание карнитина в крови. У одних больных обнаруживают уменьшенный синтез карнитина, у других — повышенную его экскрецию с мочой. Активность сывороточной КК может быть слегка повышенной. В биоптате мышцы находят накопление липидов. В некоторых случаях их накопление отмечают также в печени, сердце и почках. У некоторых больных, но далеко не у всех, эффективным оказался пероральный прием карнитина или кортикостероидов.
Карнитинпалмитилтрансферазная недостаточность. Указанная ферментная недостаточность проявляется повторяющейся миоглобинурией. Точно неизвестно, снижение активности какой карнитинпалмитинтрансферазы (КПТ) при этом происходит: КПТ-I или КПТ-II. Данная ферментная недостаточность, по-видимому, является результатом нарушения регуляции свойств патологического фермента. Большая физическая нагрузка (игра в футбол, длительный поход) может спровоцировать рабдомиолиз; однако иногда выявить провоцирующий фактор не удается. Первые признаки болезни часто появляются в детстве. В отличие от поражений мышц при нарушениях гликолиза, когда уже после кратковременных, но интенсивных физических нагрузок появляются судороги в мышцах, что заставляет больного отказываться от продолжения физической нагрузки и тем самым защитить себя, при КПТ-недостаточности боли в мышцах не возникают, пока не будут израсходованы все энергетические ресурсы мышцы и не начнется ее деструкция. Во время рабдомиолиза возникает резчайшая мышечная слабость, так что некоторым больным бывает необходима искусственная вентиляция легких. В отличие от карнитиновой недостаточности при недостаточности КПТ между приступами болезни мышечная сила сохранена, а при биопсии мышцы в ней не обнаруживают накопления липидов. Диагноз требует непосредственного исследования содержания в мышце КПТ. Лечение заключается в увеличении потребления углеводов с пищей перед физической нагрузкой или в замене в диете больного триглицеридов со средней цепью на триглицериды с длинной цепью. Однако все эти методы лечения не являются вполне удовлетворительными.
Миоаденилатдеаминазная недостаточность. Фермент аденилатдеаминаза превращает 5-аденозинмонофосфат (5-АМФ) в инозинмонофосфат (ИМФ) с высвобождением аммиака, что может играть определенную роль в регулировании содержания в мышце аденозинтрифосфата (АТФ). В 1978 г. удалось выявить группу больных с мышечными болями и непереносимостью физической нагрузки, у которых имел место дефицит изоэнзима миоаденилатдеаминазы. Недостаточность этого фермента довольно распространена и встречается примерно у 1 % населения, что может быть установлено при специальной окраске мышечных гистологических препаратов или при биохимическом исследовании мышечной ткани. При исследовании теста с нагрузкой на мышцы предплечья обнаруживается снижение образования аммиака. Со времени оригинального описания данного заболевания более четких клинических его проявлений выявить не удалось. Нередко у больных с другой нервно-мышечной патологией (поражения клеток передних рогов спинного мозга, мышечная дистрофия, миастения) также обнаруживают недостаток этого фермента. Четко клиническое значение этого нарушения не установлено.
Митохондриальные миопатии. Гетерогенная группа заболеваний, характеризующихся патологией со стороны митохондрий, обязана своим названием [ragged-red fibers («шероховатые красные волокна»)] особому виду окрашенного трихромно гистологического препарата биопсированной мышцы. Синдром Кирнса—Сейра — это спорадическое заболевание, начинающееся в детском возрасте и характеризующееся прогрессирующей экстернальной офтальмоплегией, нарушениями внутрисердечной проводимости, которая нередко приводит к полной поперечной блокаде. Отмечают также дегенерацию ретины, малый рост больных, гонадные дефекты.
Наследственное заболевание с прогрессирующей экстернальной офтальмоплегией и слабостью проксимальных мышц бывает трудно отличить от синдрома Кирнса— Сейра. Недавно был выделен еще один синдром, обозначаемый акронимом MERRF1, при котором миоклоническая форма эпилепсии сочетается с обнаруживаемыми в гистологических мышечных препаратах «шероховатыми красными волокнами». Заболевание это возникает между 1-ми 5-м десятилетиями жизни и характеризуется генерализованными судорожными припадками, миоклонусом, деменцией, потерей слуха и атаксией.
Третье заболевание из этой группы — это МЕLАS2-синдром (1 MERRF — myotonic epilepsy, ragged-red fibers (примеч. ред.). 2 MELAS — myopathy encephalopathy, lactic acidosis, stroke-like episodes (примеч. ред.)., который является медленно прогрессирующим заболеванием, характеризующимся митохондриальной миопатией, энцефалопатией, молочнокислым ацидозом, инсультоподобными эпизодами с развитием преходящих гемипарезов, гемианопсией или кортикальной слепотой, а также очаговыми или генерализованными судорожными припадками. Причина митохондриальных миопатий неизвестна, однако имеются факты в пользу того, что в семейных случаях болезнь может передаваться митохондральной, а не хромосомной ДНК.
Дата добавления: 2015-03-17; просмотров: 1321;