Гликолиз. Р-ции. Регуляция. 2 страница
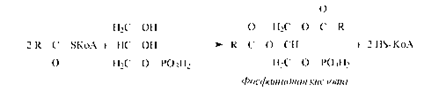
Фосфорилирование глицерина осуществляется глицеролкиназой за счет энергии АТФ. Глицерол-3-фосфат может образовываться и при восстановлении диоксиацетонфосфата.Гидролиз фосфатидной кислоты фосфатазой приводит к образованию 1,2-диацилглицерина, который, реагируя с другой молекулой ацил-КоА, образует нейтральный триацилглицерин.В слизистой кишечника триацилглицерины синтезируются из свободных кислот, моно- и диацилглицеринов, но эти процессы характерны только для слизистой оболочки кишечника. Перенос остатка жирной кислоты происходит через ацильноепроизводноеКоА.
70. Синтез кетоновых тел. Роль кетоновых тел. Биосинтез холестерина и его производных. Роль холестерина в организме.Кетоновые тела- способ транспорта ацетильной группы. К кетоновым телам относятся: ацетоацетат, 3- гидроксибутират и ацетон. Синтез ацетоацетата происходит в митохондриях печени. Затем он восстанавливается до 3- гидроксибутирата, либо расщепляется до ацетона. Далее все 3 соединения поступают в кровь и разносятся по тканям. Кетоновые тела выделяются с мочой. Кетоновые тела используются клетками всех тканей, за исключением эритроцитов и печени. Роль холестерина- в организме человека холестерин присутствует во всех клетках и тканях организма. С его участием происходят процессы выработки витамина D, стероидных гормонов коры надпочечников, женских и мужских половых гормонов, транспорт веществ через клеточные мембраны, поддерживается уровень воды в клетках. Продукты- яичный желток, печень, почки, икра, масло, сметана и др. жирные молочные продукты. Биосинтез холестерина-образование органического спирта холестерина. Синтез холестерина происходит в клетках печени, кишечнике и коже. Биосинтез холестер включает: Превращение трёх молекул активного ацетата в пятиуглеродный мевалонат. Превращение мевалоната в изопентенилпирофосфат. Образование тридцатиуглеродного изопреноида сквалена. Циклизация сквалена в ланостерин. Превращение ланостерина в холестерин.
71. Причины и типы гипо и гиперлипротеинемий. Атеросклероз,этапы атерогенеза. Функции холестерина в организме человека. Профилактика атеросклероза.Гиперлипротеинемия- повышенный уровень липидов в крови человека. Гиперлипопротеинемия I типа очень редка, резко усиливается после приема жира и снижается после строгого его ограничения. Клинические проявления возникают в возрасте до 10 лет; часто наблюдаются боли в животе, панкреатит, развитие атеросклероза не характерно. Гиперлипопротеинемия II типасоставляет около 30% случаев гиперлипопротеинемий, связана со снижением катаболизма. Характерны образования желтых пятен в области ахиллова сухожилия, сухожилий разгибателей стоп и кистей. Гиполипопротеинемия- состояние, при котором понижено содержание жиров в крови. Увеличивается риск развития атеросклероза, нарушение всасывания жиров в кишечнике, плохой аппетит. Атеросклероз- хроническое заболевание, при котором периодически обостряется процесс повреждения сосудов, характеризующийся нарушением холестеринового обмена. Этапы атерогенеза: 1.Первичное повреждение эндотелия 2.Миграция моноцитов и Т-лимфоцитов 3.Активация тромбоцитов, пролиферация гладкомышечных клеток 4. Образование жировых полосок 5.Формирование атеросклеротических бляшек. Профилактика: Значительную роль в защите от угрозы атеросклероза играют женские половые гормоны эстрогены, способствующие уменьшению содержания в крови веществ, которые принимают участие в образовании опасных наростов на стенках сосудов. Поэтому риск развития атеросклероза у женщин возрастает после климакса. Атеросклероз может развиваться и в течении 10 лет, а может и быстро. Предотвратить быстрое развитие атеросклероза можно например теплыми ваннами из отваров, применением лекарствен препаратов растит. Происхождения, с витамином С, а также употребление петрушки, мяты, чеснока. Роль холестерина- в организме человека холестерин присутствует во всех клетках и тканях организма. С его участием происходят процессы выработки витамина D, стероидных гормонов коры надпочечников, женских и мужских половых гормонов, транспорт веществ через клеточные мембраны, поддерживается уровень воды в клетках.
72. Переваривание белков в ЖКТПереваривание пищи начинается в ротовой полости,где ферменты L и B(бетта) амилазы отщепляют углеводные компаненты от сложных белков.Далее в желудке белки подвергаются действию HCL и пепсина.HCL выполняет следующие функции:-обеззараживает пищу,активирует профермент пепсиногена,превращая его в активный пепсин.,способствует частичной денатурации белков,делая их более доступными для пепсина.,обеспечивает оптимальный pH для действия пепсина.Пепсин синтезируется главными клетками желудочных желез в виде профермента пепсиногена.В полости желудка отщепляется 47 аминокислотных остатков с N конца и олбразуется активный пепсин,который далее сам активирует предшественник. Пепсин яв-сяэндопептидазой и расщепляют белки на более короткие момент.Далее пища поступает в тонкий кишечник,где нейтрализуется при помощи бикарбонатов,содержащихся в панкретаическом соке.В верхнем отделе тонкого кишечника на белки воздействует фермент трипсин, эластаза, химотрипсин.Предшественники этих ферментов синтезируются в поджелудочной железе и секретируются в просвет кишечника.Энтероциты секретируют фермент энтеропептидазу,активирующюю предшественники.В результате действия фермента образуются короткие полипетиды,на которые воздействует фермент карбоксидипептидаза,отщепляющий аминокислоты с «С» конца.Окончательное расщепление до аминокислот обеспечивает аминодипептидаза(отщепляет остатки с N конца). Всасывание и транспорт амин.к-т.Всас-е осущ-ся в тонк.кишкеmax концентр-я аминк-т в крови достигается ч/з 30-50 мин.после потреб-я белка.Энтероциты имеют 4 системы источн-ка для трансп-та аминк-т: 1.нейтральных ак-т,2.основных ак-т,3.кислыхак-т.4.для транспорта глицина и аминк-т(пролина и гидроксипролина).D-аминк-ты всасываются при помощи прост.диффузии.Работапереносщиков L-аминок-т требует затрат E и сопряж-но с деят-тьюNa+ K+ АТФазы. Мех-м напоминает всасывание глюкозы. Пример: симпорт с Na,затем удал-е Na из кл-киактивн.транспортом. В процессе уч-ет также витамин В6.Универсальным мех-змомтрансп-та аминк-т явл-ся гамма-глутамильныйцикл.В нем участ-ет 6 ферментов(1-мембрансвязанный,а остальные в цитозоли) и 3-пептидглутатион(гамма-глутанилцистеинилглицин). Ключевым ферментом яв-ся гамма-глутамилтранф-за, которая катализирует перенос глутамин-го остатка от глутатиола на переносимую к-ту.Образ-сяглутамил-аминок-та и цистиниилглицин.Они перенос-ся в цит-му,гдерасщ-ся ферм-ом глутамил-аминотрансферазой до свободной аминок-ты и 5-оксопролина. Одновременно происходит гидролиз дипептинаглутатион в ходе 3х послед-х р-ций,при этом затраг-ся 3 малекулы АТФ на 1 мал-луглутациона.В тонкой кишке в малом кол-ве могут всас-сяолигопептиды и некоторые нативныебелки.Этим об-сявозн-е аллерг-х р-ций поскольку антигенами могут быть только интактные белки. Всасываемыеаминок-ты попадают в портальный кровоток->печень->общ.кровоток.Наиболее интенсивно аминк-ты поглащ-ся печенью и почками,ткани мозга избирательно поглащают метионин, глетидин, глицин, оргинин,глутамин,тирозин(быстро);а так же медлено:лейцин,лизин,пролин.
73. Общие пути катаболизма аминокислот. Значение реакций дезаминирования, трансаминирования, декарбоксилирования. Диагностическое знаачение трансамилаз в сыворотке крови. Общие пути обмена аминокислот.Общие пути превращения аминокислот включают реакции дезаминирования, трансаминирования, декарбоксилирования, биосинтеза. Дезаминирование аминок-т. Первая стадия является ферментативной и завершается образованием неустойчивого промежуточного продукта, который на второй стадии в присутствии воды распадается на аммиак и α-кетокислоту. Трансаминирование аминокислот- реакции межмолекулярного переноса аминогруппы от аминокислоты на α-кетокислоту без промежуточного образования аммиака. Реакции являются обратимыми и универсальными для всех живых организмов, протекают при участии специфических ферментов (аминофераз). Специфичность трансаминаз обеспечивается белковым компонентом. Ферменты трансаминирования катализируют перенос NH2-группы не на α-кетокислоту, а сначала на кофермент перидоксальфосфат. Образовавшееся промежуточное соединение подвергается внутримолекулярным превращениям, приводящим к освобождению α-кетокислоты и пиридоксаминфосфата; последний на второй стадии реакции реагирует с любой другой α-кетокислотой, это приводит к синтезу новой аминок-ты и освобождениюпиридоксальфосфата. Декарбоксилирование аминокислот. Процесс отщепления карбоксильной группы аминокислот в виде СО2. Образующиеся продукты р-ции – биогенные амины – оказывают сильное фармакологическое действие на множество физиологических функций человека и животных. В живых организмах 4 типа декарбоксилирования аминокислот: 1. α-Декарбоксилирование, характерное для тканей животных; 2. ω-Декарбоксилирование, свойственное микроорганизмам. 3. Декарбоксилирование, связанное с реакцией трансаминирования. 4. Декарбоксилирование, связанное с реакцией конденсации двух молекул.
74. Окислительный катаболизм аминокислот: возможные пути расщепления углеводного скелета, утилизация аминного азота.Углеродные скелеты аминок-т обр-ся из продуктов обмена, аминогруппы вводятся путем прямого аминирования или трансаминирования, лишь немногие из аминок-т обр-ся в рез-те прямого аминирования свободными ионами NH4+. Из свободных аминокислот в цитоплазме количественно преобладает глутаминовая к-та(более 50% пула) Расщепление амин-т включает 2 типа реакций 1)Связанные с удалением и дальнейшим превращением аминогруппы (дезаминирование, переаминирование, включается в состав мочевины).2)превращение углеродного скелета. Превращение углер. скелета в аэробных условиях приводят к соединениям, включающимся в ЦТК.Удаление аминогруппы происходит переаминированием.Аланин и аспартат обр-ся путем трансаминирования соответственно из пирувата и аксалоацетата.
Тирозин обр-ся при гидроксилировании фенилаланина, цистеин синтез-ся из метионина и серина в сложн. последовательности реакций с образованием в качестве промежуточных продуктов S-аденозилметионина и цистатеонина. Углеродн.скелет серина происходит от 3-фосфоглицерата. Серин явл-ся предшественником глицина;-углеродный атом серина переносится тетрагидропалатом,явл-ся переносчиком одноуглеродных групп. Конечные продукты, образующимся в рез-те катаболизма амин-ты делят на 3 гр: 1)Глюкогенные (аланин,аргинин,аспарагин,аспарагин.к-та,валин,глутамин.к-та, глицин, гистидин, метионин,пролин,серин,треонин,триптофан,цистеин) 2)Кетогенные (образуют кетоновые тела) лейцин,лизин,триптофан 3)Смешанные (кетогенные и глюкогенные)тирозин,фенилаланин. Сера с серосодержащих амин-т передается на др.серосодержащие амин-ты при их образовании с цистеина на цистеин или с метионина на метионин или отщепляются в виде SO4,из которого затем в цепи реакций возникают SO4 ,лишняя S выводится из организма с мочой в 3х формах: а)неорганические сульфаты 80%, б)сера в составе эфиров, в)органическая S в составе амин-т.Ароматические циклы расщепляются при действии оксидаз или передаются вновь синтезируемой амин-те. Повышение уровня трансаминаз в сыворотке крови отмечено при некоторых заболеваниях мышц, при обширных травмах и прогрессивной мышечной дистрофии.
75. Обмен одноуглеродных групп как способ углеродного скелета при биосинтезе аминокислот и нуклеотидов. Обмен серина, глицина и треонина.Углеродные скелеты аминок-т обр-ся из продуктов обмена, аминогруппы вводятся путем прямого аминирования или трансаминирования, лишь немногие из аминок-т обр-ся в рез-те прямого аминирования свободными ионами NH4+.В первичной ассимиляции аммиака участвуют L-глутаматдегидрогеназа,L-аланиндегидрогенеаза, кот.осуществ-ют восстановительное аминирование кетокислот без участия АТФ. Обр-ие глутамина из глутамата, катализируется глутаминсинтетазой при участии АТФ,фермент активен даже при низк.конц-ях NH4+.Большенство др.аминокислот получают аминогруппу в рез-те трансаминирования. Из свободных аминокислот в цитоплазме количественно преобладает глутаминовая к-та(более 50% пула).Аланин и аспартат обр-ся путем трансаминирования соответственно из пирувата и аксалоацетата. Тирозин обр-ся при гидроксилировании фенилаланина, цистеин синтез-ся из метионина и серина в сложн. последовательности реакций с образованием в качестве промежуточных продуктов S-аденозилметионина и цистатеонина.Углеродн.скелет серина происходит от 3-фосфоглицерата.Серин явл-ся предшественником глицина;-углеродный атом серина переносится тетрагидропалатом,явл-ся переносчиком одноуглеродных групп (метильная, формильная,гидроксиметильная). Аллостерич. регуляция биосинтеза аминокислот осущ.по принципу обратной связи, при этом регуляторным явл-ся первый фермент цепи реакций.
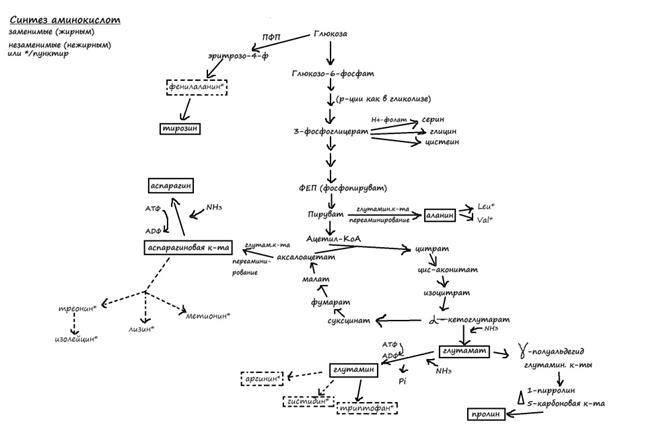
76. Обмен фенилаланина и тирозина. Фенилкетонурия. Фенилаланин – незаменимая к-та. Основной его путь превращения начинается с его гидроксилирования в тирозин. Реакция катализируется специфической фенилаланин-4-монооксигеназой, которая в качестве кофермента содержит тетрагидробиоптерин. Блокирование этой реакции приводит к развитию тяжелой наследственной болезни – фенилкетонурии (фенилпировиноградная олигофрения). В процессе трансаминирования тирозин превращается в n-оксифенилпировиноградную кислоту, которая под действием специфической оксидазы подвергается окислению, декарбоксилированию, гидроксилированию и внутримолекулярному перемещению боковой цепи с образованием гомогентизиновой кислоты; эта реакция требует присутствия аскорбиновой к-ты, роль которой пока не выяснена. Дальнейшее превращение гомогентизиновой кислоты в малеилацетоуксусную к-ту катализируется оксидазой гомогентизиновой кислоты. Малеилацетоуксусная к-та под действием специфической изомеразы в присутствии глутатиона превращается в фумарилацетоуксусную к-ту, подвергающуюся гидролизу с образованием фумаровой и ацетоуксусной кислот. Фенилаланин и тирозин являются также предшественниками меланинов. Фенилкетонурия - наследственное заболевание, в основе которого лежит аномалия аминокислотного обмена вследствие отсутствия или резкого снижения активности фермента фенилаланингидроксилазы. Фермент сохраняет только около 5% активности, в связи с чем нарушается обмен фенилаланина и вследствие этого – тирозина, триптофана и др., накапливаются промежуточные продукты обмена – фенилэтиламин, фенилпировиноградная кислота и др. и возникает дефицит метаболитов, необходимых для нормального функционирования организма. Фенилкетонурия проявляется выраженной олигофренией (идиотией или имбецильностью). Диагностируется в первые дни жизни ребёнка с помощью микробиологических или биохимических методов.
77. Метаболизм метионина. Метионин – незаменимая аминок-та, которая не синтезируется в орг-зме ч-ка. Поэтому метионин должен постоянно поступать в орг-зм вместе с пищей. Метионин входит в состав белка. В метаболизме роль метионина связана с тем, что она содержит подвижную метильную группу (-СНз), которая может передаваться на другие соединения. Способностью метионина отдавать метильную группу обусловлен его липотропный эффект (удаление из печени избытка жира). Отдавая подвижную метильную группу, метионин способствует синтезу холина, с недостаточным образованием которого связаны нарушение синтеза фосфолипидов из жиров и отложение в печени нейтрального жира. Метионин участвует в синтезе адреналина, креатина и других биологически важных соединений; активирует действие гормонов, витаминов (В 12 , аскорбиновой и фолиевой кислот), ферментов. Если нарушается метаболизм, то организм человека «теряет равновесие», что неотвратимо приводит к расстройствам и заболеваниям. Путем метилирования (отдачи -CH3-группы) метионин обезвреживает токсичные продукты. Применяют метионин для лечения и предупреждения заболеваний и токсических поражений печени, а также при хроническом алкоголизме, сахарном диабете и др. Эффект более выражен при жировой инфильтрации клеток печени. При вирусном гепатите применять метионин не рекомендуется. Метионин назначают для лечения дистрофии, возникающей в результате белковой недостаточности у детей и взрослых после дизентерии и других хронических инфекционных заболеваний. Метионин способствует снижению содержания холестерина в крови, уменьшению отложения жира в печени и улучшению функции печени, может оказывать умеренное антидепрессивное действие.
78. Метаболизм гистидина. Дезаминирование гистидина происходит в печени и коже под действием фермента гистидазы с образованием уроканиновой к-ты, которая затем в печени превращается в имидазолонпропионовую к-ту под действием уроканиназы. Дальнейшее превращение в ходе серии р-ций имидазолонпропионовой кислоты приводит к образованию аммиака, глутамата и одноуглеродного фрагмента, соединённого с тетрагидофолиевой кислотой. Реакция декарбоксилирования гистидина имеет большое физиологическое значение, так как является источником образования биологически активного вещества — гистамина, который играет важную роль в процессе воспаления и развития некоторых аллергических р-ций. Декарбоксилирование происходит большей частью в тучных клетках соединительной ткани практически всех органов. Эта р-ция протекает при участии фермента гистидиндекарбоксилазы. Известно связанное с дефектом гистидиназы наследственное заболевание гистидинемия, при котором характерно повышенное содержание гистидина в тканях и задержка умственного и физического развития.
79. Биогенные амины.Явл. продуктами декарбоксилирования аминок-т и обладают повышенной биолог. активностью. К группе относятся многие нейромедиаторы: 1)гамма-аминомаслянная кислота-образ-ся при декарбоксилировании глутаминовой к-ты (ферм. – глутамандекарбоксилаза, коферм.- передоксальфосфат). Основное место синтеза-ткань головного мозга, главный тормозный медиатор в ЦНС, вызывает гиперполяризацию постсинаптической мембраны вследствие обратного транспорта ионов CL и накопл-е в клетке Са. Поэтому сигнал от возбуждающегося нерва не достигает порогового уровня. Распад гамма-аминомаслянной к-ты происходит в рез-те переаминирования с альфа-кетоглутарата, т.к. образуется сукцинат и глутамат. Тормозным медиатором в спинном мозге и в стволе-глицин(антагонист стрихнин). 2)Гистамин образ-ся при декарбоксилировании гистидина, катализируемого специфической декарбоксилазой. Основное место синтеза –тучные клетки, в которых он находится в виде белково-гистаминового комплекса. Освобожд-ся при действии спец. факторов (либераторов). Гистамин активирует секрецию пепсиногена и НCL в слизистой желудка и является сильным сосудорасширяющим агентом и медиатором аллергических реакций. В большых кол-вах освобождается из ДЕПО при травматическом шоке и зоне воспаления, распадается под действием диаминооксидазы. 3)Серотонин образ-ся из триптофана, при гидроксилировании, (фермент триптофан-5-моно-оксигеназа), (кофермент-тетрагидроптередин).С последующим декарбоксилированием образ-ся с нейронами гипоталамуса и ствола мозга и явл. медиатором этих нейронов. Сильный сосудосуживающий агент, повышает свертываемость крови. Разруш-ся моноаминооксидазой, образ-ся оксиэндолилуксусная к-та, которая выводится из орг-ма с мочой. 4)Дофамин производное тирозина. Под действием тирозиназы тирозин гидроксилируется до 3,4-диоксифенилаланина(ДОФА) . ДОФА декарбоксилир-ся до дофамина (в почках, надпочечниках, в симпатических ганглиях и нервах).Медиатор ингибированного типа одного из проводящих путей (в черной субстанции верхнего отдела ствола мозга и в полосатом теле явл-ся предшественником меланина, норадреналина и адреналина. 5)Норадреналин Медиатор в постганглионарных волокнах симпатической нервной системы, активирует аденилациназу. Это приводит к увеличению уровня синтеза циклической АМФ и активации протеинкиназ, что повышает активность ферментов в клетке. Образ-ся при гидроксилировании ДОФА при помощи фермента дофамин-бета-монооксикеназы, также как и адреналин явл-ся гормоном надпочечников. 6)Адреналин образуется в рез-те азотметилированного норадреналина, (фермент-фенилэтанолфолин-азот-метилтрансфераза),усиливает мобилизацию гликогена. 7)Таурин образ-ся из цистеина, синтезируется во многих органах и тканях, выполняет медиаторную ф-цию на уровне синапсов. Участвует в образовании коньюгированных жирных к-т.
81. Общие принципы обмена аминок-т. Нарушение обмена белков и аминок-т .Белковая недостат-ть развив-ся у ч-ка как при полном, так и при частичном голодании, так и при приеме однообразных белков в питании. Когда в диете преоблад. растит. белки - биологич.ценность которых ниже животных белков, результат-отриц. азотистый баланс (кол-во поступающего азота меньше кол-ва выводимого из орг-ма). Гипопротеинемии (понижение концентрации белков в сыворотке до 30-50гр/л. Нарушение коллоидно-осмотического и водно-солевого обмена(развитие отеков). При тяжелых формах пицеварительной дистрофии наблюдается поражение печени, остановка роста, резкое снижение иммуного столбца, отечность, атомия м-ц. При белковой недостаточности резко понижается интенсивность процессов дезаминирования, трансаминирование и биосинтез аминок-т, а также активность цикла мочевины в печени из-за недостаточного синтеза нужных ферментов, следует аминоацидурия до 10-20 г/сутки (в норме 1г в сутки), повышение концентрации свободных аминок-т в крови и снижение экскреции мочевины .Увелич-ся уровень распада белков. Гипераминоацидурия делится на 1)почечную (связана с приобретенным или врожденным дефектом реабсорбции аминок-т в почки ); 2)Внепочечную (связана с повышенной концентрацией аминок-т в крови).Причины почечной аминоацидурии служат -Хронич. нефрит, нефрозы, наследств.заболевания такие как цистиноз (врожден.нарушение реабсорбции почти всех аминокислот-повыш-е в 5-10 раз экскреции всех аминок-тв 20-30 раз цистина и цистеина, отложении цистина в клетках РЭС, селезенки, печени и роговицы глаза) и цистинурия( наруш-е реабсорбции и повышения выведения мочой цистина, аргинина, лизина, орнитина в 50 раз, повыш-ся вероятность образования камней в почках.Некоторые энзимопатии
| Заболевание | фермент |
| альбинизм | Тирозин-3-монооксигеназа |
| алкаптонурия | Гомогентизат-1,2-диоксигеназа |
| оргининосукцинатацидемия | Оргинино-сукцинат лиаза |
| гомоцистинурия | Цистотиамин-бета-синтаза |
| Болезнь Клинового сиропа(лейциноз) | Дегидрогеназа альфа-кетокислот с развлетвленной цепью |
| Фенилкетонурия | Фенилаланин-4-монооксигеназа |
| гипервалинемия | Валинтрансаминаза |
Аминокислоты широко используются в современной фармакологии. Являясь не только структурными элементами белков и других соединений, они имеют большое значение. Некоторые из них выступают в качестве нейромедиаторных веществ (глутаминовая, аспарагиновая кислоты, глицин, таурин, Ag -аминомасляная кислота и др.). Фенилаланин и тирозин являются предшественниками в биосинтезе дофамина, норадреналина, адреналина; триптофан — предшественником серотонина; гистидин — предшественником гистамина. Производными аминокислот являются энкефалины, эндорфины, динорфины и другие нейропептиды, а также высвобождающие факторы(рилизинг-факторы) гипоталамуса, гормоны гипофиза и т. д. Некоторые аминокислоты (глутаминовая, Ag -аминомасляная, метионин, глицин и др.) нашли самостоятельное применение в качестве лекарственных средств. Расширяется круг новых лекарственных препаратов, синтезируемых с использованием остатков аминокислот (см. Даларгин, Каптоприл, Тимоген и др.). Специальное значение имеют смеси аминокислот, используемые в качестве средств для парентерального питания.
83. Реутилизация пуриновых оснований. Гиперурикемия. Синдром Леша-Нихана. Подагра, причины и сущность заболевания принципы лечения.Реутилизация пуриновых оснований- процесс повторного их использования. Актуален в быстрорастущих тканях, когда активно идет процесс синтеза нуклеин.к-т и недопустима потеря их предшественников. Сущ-ет 2 способа реутилизации:1способ- заключается в присоединении рибозо-5-фосфата к свобод.основаниям гуанину, аденину или гипоксантину с образованием АМФ, ГМФ или ИМФ

2 способ: реутилизируются пуриновые рибонуклеозиды или дезоксирибонуклеозиды. Для этого сущ-ет фермент аденозинкиназа и дезоксицитидинкиназа. Гиперурекимия-повышенное содержание моч.к-ты в крови.
Причиной такого повышения яв-ся 2 фактора: нарушение выведения почками мочевой кислоты- почечная гиперурекимия; избыточное её образование-обменная гиперурекимия. Гиперурекимия вызывается: ускоренным образованием моч.к-ты из-за участия пурина в обмене в-в; из-за ослабленной работы почек; из-за повышенного содержания фруктозы в пище. Гиперурекимия также вызывает голодание и потребление высококалорийной пищи. Одним из способов лечения яв-ся потребление пищевой соды-она понижает содержание кислотной мочи. Синдром Леша-Нихана.Сопровождается проявлением подагры. Основной фактор-дефект гипоксантина, гуанига, фосфорибозилтрансферазы,катализир.превращением гипоксантина и гуанина в инозинмонофосфат и ГМФ. При этом гуанин и гипоксантин превращаются в мочев.к-ту и не испол-ся повторно в синтезе мононуклеотидов. Заболев-е отмечается у лиц мужского пола. Характерным признаком болезни являются аутоагрессивные действия. Диагноз синдрома Лёша-Нихена ставится по трём элементам: повышенная продукция мочевой к-ты, неврологическая дисфункция, поведенческие нарушения. Подозрения могут возникнуть из-за задержки развития, сопровождающейся гиперурикемией. Также, возможно образование камней в почках (нефролитиаз) или наличие крови в моче (гематурия). Зачастую подозрения на синдром Лёша-Нихена возникают с появлением наносимых самому себе ранений у больного. Подагра -это болезнь обмена веществ, при котором соли мочевой кислоты(ураты) откладываются в суставах. Обусловлена 3 факторами: Повышением синтеза мочев к-ты; Снижением содержания в плазме урат связывающих белков; Уменьшением ренальной фильтрации и соответственно замедлением выведения моч к-ты. В развитии подагры 4 стадии: 1)Бессимптомная гиперурекимия - повышенное содержание мочевой кислоты в крови без каких-либо признаков отложения кристаллов . 2)Острый приступ - развивается обычно через несколько лет бессимптомной гиперурикемии; 3)Стадия межприступной подагры-чередование 1 и 2 стадии. Приступы становятся более тяжелыми, захватывают «новые» суставы. Наблюдается воспаление связок, суставных сумок, часто образуются единичные, обычно безболезненные тофусы. 4)Хроническая тофусная подагра-характеризуется наличием тофусов, хронического артрита, поражением почек, нефролитиазом. Тофусы локализуются обычно подкожно или внутрикожно в области пальцев кистей и стоп, коленных и локтевых суставов, на ушных раковинах. Над тофусами кожа может изъязвляться с выделением содержимого в виде пастообразной белой массы. При подагре возрастает частота сахарного диабета, атеросклеротического поражения сосудов. Лечение:противовоспалительные препараты (индометацин,аллопуринол);снизить содержание мочевой кислоты в крови;потребление адекватного объема жидкости;исключение приема лекарственных препаратов, повышающих уровень мочевой кислоты в крови (в первую очередь диуретиков); Диетотерапия- исключение из рациона пищевых продуктов, содержащих большое количество пуринов!
84. Биосинтез и распад пиримидиновых нуклеотидов: этапы, регуляция. Оротацидурия. Биосинтез. Исходным соединением в синтезе пиримидиновых нукл-в явл. карбамоилфосфат, который конденсируясь с аспарагиновой кислотой образует карбамоил-аспарагиновую к-ту, возникающую при замыкании цикла и последовательном окислении оротат реагирует с фофорибозилпирофосфатом, образуя оротидиловую к-ту. Декарбоксилирование оротидилата дает уридилат, который далее превращается в уридил-3-фосфат. Присоединение аминогруппы от глутамина приводит к образованию цитидин-3-фосфата.Скорость биосинтетических реакций регулируется на уровне первой реакции. Регулятором служит фермент аспартаттранскарбамоилаза. Тимидиновые нукл-ды образуются из дезоксидилмонофосфата при помощи тимидилатсинтетазы. Распад. Распад пиримидинов протекает по одному из нескольких механизмов, обнаруженных у разных видов организмов. Например, у человека реализуется такой механизм, которыйвключает дефосфорилирование и отщепление углеводного компонента от нуклеотидов с образованием тимина или урацила; затем происходит восстановление тимина или урацила с образованием полностью гидрированного гетероцикла. Расщепление цитозина происходит аналогично после того, как он дезаминируется в урацил. Раскрытие кольца в промежуточном продукте приводит к образованию карбомоил-аланина, который далее гидролизуется до СО2, NH3 и β-аланина. Все продукты либо выводятся из организма, либо повторно утилизируются в других метаболических процессах. Например, β-аланин может повторно быть использован в биосинтезе кофермента А.Нарушение обмена пиримидиновых нуклеотидов проявляется в виде наследственного заболевания-оротацидурии.С мочой выделяется оротовая к-та в кол-ве превышающем норму. Причина: дефицит дегидрогеназы. Происходит накопление оротата и в результате наблюдается недостаточность пиримидина=>отставание физического и умственного развития.Лечение: устранение пиримидиновой недостаточности путём введения уридина.
Дата добавления: 2015-03-11; просмотров: 1039;