Сроки прорезывания зубов
| Схема расположения зубов (номера показывают порядок появления) | Зубы в порядке появления | Средний срок появления (месяцы) |
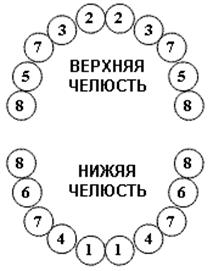
| Нижние центральные резцы (1) | 6 – 7 |
| Верхние центральные резцы (2) | 8 – 9 | |
| Верхние боковые резцы (3) | 9 – 11 | |
| Нижние боковые резцы (4) | 11 – 13 | |
| Верхние первые моляры (5) | 12 – 15 | |
| Нижние первые моляры (6) | 12 – 15 | |
| Клыки (7) | 18 – 20 | |
| Вторые моляры (8) | 20 – 30 |
У многих детей наблюдается нарушение последовательности и сроков прорезывания. Парность появления зубов также не всегда соблюдается. Сроки появления зубов связаны как с врожденными особенностями организма, так и с общим состоянием здоровья ребенка. Причиной задержки прорезывания зубов может быть рахит или другие нарушения обменных процессов.
Долженствующее количество зубов для определенного возраста можно рассчитать по формулам.
Для молочных зубов: х = n – 4, где х – количество зубов, n – возраст ребенка (в месяцах).
Для постоянных зубов: х = 4n – 20, где х – количество зубов, n – возраст ребенка (в годах).
ОЦЕНКА УРОВНЯ БИОЛОИЧЕКОЙ ЗРЕЛОСТИ ПО ВТОРИЧНЫМ ПОЛОВЫМ ПРИЗНАКАМ
(см. главу «Половое развитие детей»)
СЕМИОТИКА НАРУШЕНИЯ РОСТА
СЕМИОТИКА НИЗКОГО РОСТА
Задержка роста является частой причиной обращаемости к эндокринологу: около 3 % детей имеют выраженную задержку роста. При этом дефицит гормона роста как причина низкорослости выявляется не более чем у 8,5 % из них. У остальных детей наиболее часто выявляются конституциональные особенности роста и развития, реже — дефицит других анаболических гормонов, тяжелые соматические заболевания, генетические и хромосомные заболевания.
Принято различать эндокриннозависимые и эндокринноно-зависимые формы задержки роста.
Эндокриннозависимые варианты задержки роста. Наиболее тяжелые нарушения процессов роста наблюдаются при патологии эндокринной системы. Известно, что многие гормоны (соматотропный, тиреоидные, половые, глюкокортикоиды, инсулин и др.) непосредственно или пермиссивно участвуют в процессах роста. Наиболее выраженный ростовой эффект дает соматотропный гормон (СТГ, соматотропин, гормон роста). СТГ — это белок, состоящий из 191 аминокислотного остатка и кодируемый геном 17-й хромосомы. По структуре гормон роста на 85 % гомологичен хорионическому соматомаммотропину и на 26 % — пролактину. В отличие от других гормонов гипофиза он относительно видоспецифичен: в отношении человека эффективен только гормон роста приматов. СТГ, применяющийся для лечения детей с его недостаточностью, в настоящее время синтезируется с помощью рекомбйнантной ДНК.
В организме СТГ синтезируется и депонируется соматотрофами аденогипофиза. Секреция СТГ имеет пульсирующий характер с выраженным суточным ритмом. Основное количество СТГ образуется в ночное время, в начале глубокого сна, что особенно выражено в детстве. Секреция соматотропина стимулируется СТГ-рилизинг-гормоном (соматолиберином) и тормозится СТГ-ингибирующим фактором (соматостатином). Их эффекты опосредуются гипоталамическими нейротрансмиттерами (дофамином, норадреналином, серотонином и др.). Поэтому психический стресс, и особенно дистресс, физические перегрузки, прием многих лекарственных средств изменяют оптимальную и синхронизированную активность нейротрансмиттеров гипоталамуса, вследствие чего нарушается секреция соматостатина и соматолиберина и в результате — секреция СТГ. СТГ является основным гормоном, стимулирующим линейный рост. Он способствует росту костей в длину, росту и дифференцировке внутренних органов, развитию мышц. СТГ стимулирует липолиз (снижая массу жировой ткани), гликонеогенез, синтез белка, увеличивает мышечную массу и силу, всасывание кальция и фосфора в кишечнике и оссификацию костей. Показано усиление сократимости миокарда и скорости клубочковой фильтрации под влиянием соматотропина.
Ростостимулирующие эффекты СТГ опосредуются через соматомедины или инсулиноподобные факторы роста (ИФР-1, ИФР-2), которые синтезируются главным образом в печени и почках под влиянием СТГ. ИФР стимулируют пролиферацию и дифференцировку хондробластов, обеспечивая рост кости в длину. Соматомедины циркулируют в плазме в связанном со специфическими белками-носителями виде. Известно 6 классов этих белков. Наиболее важным из них является ИФРСБ-3, который связывает около 90 % всех ИФР у детей и взрослых. Уровень ИФР-1 и ИФРСБ-3 отражает суммарную секрецию СТГ.
Стимулирующее влияние на секрецию СТГ оказывают тиреоидные и половые гормоны, вазопрессин, адренокортикотропный, меланоцитостимулирующий гормоны. Глюкокортикоиды при острой нагрузке оказывают стимулирующее действие; при длительном хроническом избытке гормона секреция СТГ практически полностью подавляется.
Тиреоидные гормоны в физиологических количествах дают значительный анаболический эффект. В отличие от СТГ эти гормоны в большей степени влияют на дифференцировку (созревание) тканей, прежде всего костной. В то же время тиреоидные гормоны, активно влияя на уровень гормона роста, ускоряют и линейный рост ребенка.
Инсулин играет важную роль в регуляции процессов роста, так как, с одной стороны, обеспечивает анаболические процессы энергетически, с другой — непосредственно усиливает синтез белка.
Половые гормоны обладают мощным анаболическим действием, ускоряя как линейный рост, так и дифференцировку костей скелета. Однако следует помнить, что ростовой эффект половых гормонов осуществляется лишь в присутствии СТГ. Влияние эстрогенов на процессы роста доказано в настоящее время не только у девочек, но и у мальчиков на основе сверхчувствительных методов их определения. Минимальные уровни эстрогенов у детей обоего пола стимулируют ростовой скачок, высокие уровни ускоряют эпифизарное созревание и прекращают линейный рост.
Глюкокортикоиды, усиливая процессы гликонеогенеза, оказывают выраженное катаболическое действие. Отрицательное влияние на процессы роста оказывает кортизол в больших дозах, потому что активно тормозит выделение соматотропина.
Таким образом, задержка роста и отставание костного возраста являются симптомом многих эндокринных заболеваний, для которых характерен дефицит анаболических или избыток катаболических гормонов.
Церебрально-гипофизарный нанизм (ЦГН) характеризуется выпадением функций всех тропных гормонов (пангипопитуитаризм). Различаются идиопатический и органический варианты заболевания. При идиопатическом варианте ЦГН признаки органического поражения ЦНС отсутствуют, патологический процесс формируется на уровне гипоталамических структур. Встречается у 1 из 10—15 тыс. детей, у мальчиков в 2—4 раза чаще, чем у девочек. Выявлена определенная связь заболевания с рождением ребенка в ягодичном предлежании, с помощью щипцов, с кровотечением в родах. Все это свидетельствует о роли родовой травмы и гипоксии в генезе ЦГН. Клиническая картина заболевания обусловлена дефицитом тройных гормонов и нарушением в силу этого функции эндокринных желез. При этом доминируют симптомы дефицита СТГ, то есть имеет место выраженная пропорциональная задержка роста. При отсутствии лечения рост взрослых больных не превышает 120 см у женщин и 130 см у мужчин. При рождении и в первые месяцы жизни физическое развитие детей с ЦГН не отличается от такового у здоровых детей. Задержка роста становится заметной на 2-м году жизни. Постепенно темпы роста снижаются, и после 4 лет жизни дети прибавляют в год не более 2—3 см. Костный возраст значительно отстает от хронологического. Помимо задержки роста у детей с дефицитом СТГ имеется склонность к гипогликемическим состояниям (снижены процессы гликогенолиза). Гипогликемия у некоторых детей может быть первым признаком заболевания и нередко выявляется уже в период новорожденности. Дефицит тиреотропного гормона (ТТГ) у больных с ЦГН является причиной гипотиреоза, что определяет комплекс характерных симптомов: психическая вялость, сухость кожных покровов, брадикардия, гипотония, запоры, позднее появление и поздняя смена зубов. Выраженный дефицит ТТГ еще более ухудшает процессы роста и дифференцировки костей скелета у больных с ЦГН. Дефицит гонадотропных гормонов (ГТГ) является причиной развития гипогонадизма. У части мальчиков с ЦГН уже при рождении имеются признаки внутриутробного дефицита ГТГ: крипторхизм и микрофаллос. В дальнейшем у всех больных выявляются симптомы тяжелого гипогонадизма: вторичные половые признаки отсутствуют, зоны роста остаются открытыми. Выраженный дефицит половых гормонов и отсутствие вследствие этого пубертатного скачка в росте у таких детей еще более усугубляют задержку роста. У большинства больных ЦГН наблюдаются дефицит АКТГ и гипокортицизм, однако вне стрессовых ситуаций симптомы гипокортицизма у больных, как правило, не проявляются. На фоне терапии тиреоидными и анаболическими препаратами потребность в глюкокортикоидах возрастает и могут выявляться симптомы надпочечниковой недостаточности, чаще — в ответ на стрессовую ситуацию. При органическом варианте ЦГН может иметь место повреждение гипоталамо-гипофизарной системы вследствие врожденных дефектов (аплазия или гипоплазия, аневризма) или деструктивных повреждений: в перинатальный период — родовая травма, гипоксия, геморрагический инфаркт, травматические повреждения головы, переломы основания черепа. Однако наиболее часто у таких больных выявляется врожденная опухоль — краниофарингиома, могут быть и другие новообразования (гистиоцитоз X, герминома, глиома зрительного тракта, аденомы гипофиза). Помимо задержки роста у больных с органическим вариантом ЦГН обращают на себя внимание выраженная неврологическая симптоматика, признаки повышения внутричерепного давления, ограничение полей зрения, атрофия и отек дисков зрительного нерва, параличи и парезы мышц, иннервируемых черепными нервами.
Облучение опухолей ЦНС, глаз и среднего уха, так же как и облучение черепа при остром лейкозе, может обусловить гипоталамо-гипофизарные нарушения. При этом чаще всего развивается недостаточность СТГ, но возможна и недостаточность тиреотропного гормона гипофиза, АКТГ и гонадотропинов. Первые клинические признаки могут появляться спустя длительное время после облучения.
Изолированный дефицит СТГ. При изолированном дефиците СТГ другие тропные гормоны выделяются в нормальных количествах, в связи с чем наблюдается более благоприятное течение заболевания: рост взрослых больных несколько выше, чем при ЦГН (у женщин — 125 см, у мужчин — 145 см), симптомов гипотиреоза нет, половое созревание наступает обычно на 2—4 года позже, но протекает нормально, больные, как правило, фертильны. Костный возраст отстает от хронологического, но дифференцировка костей скелета нарушается в меньшей степени, чем при ЦГН. По окончании пубертатного периода зоны роста у больных закрываются. В настоящее время известны еще три варианта изолированного дефицита СТГ: частичный дефицит гормона роста, селективный дефицит гормона роста и психологическая карликовость.
Частичный дефицит СТГ встречается примерно у 10 % больных с изолированным дефицитом СТГ. Этот вариант заболевания характеризуется неполным выпадением функций гормона роста и более легким клиническим течением заболевания.
Нейросекреторная дисфункция характеризуется нарушением регуляции синтеза СТГ и снижением в силу этого спонтанной секреции соматотропина. При этом может нарушаться лишь один из регуляторных механизмов (катехоламиновый, серотониновый или допаминовый).
Психологическая (депривационная) карликовость может иметь место у детей из неблагополучных семей. У них развиваются выраженная задержка роста, отставание костного возраста, психического развития, доказано наличие дефицита гормона роста. Для обделенных душевным теплом детей характерны извращенный аппетит или прожорливость, недержание мочи и кала, бессонница, судорожный крик и внезапные вспышки гнева. Они бывают чрезмерно пассивными или агрессивными, а их интеллект находится на нижней границе нормы или снижен. При изоляции этих детей от неблагоприятных условий уровень СТГ самостоятельно восстанавливается, дети начинают расти, однако отставание интеллектуального развития остается на всю жизнь.
Редкий вариант задержки роста— синдром Ларона — обусловлен дефектом рецепторов СТГ. Клиническая картина у больных с подобным синдромом идентична клинической картине изолированного дефицита СТГ. Однако уровень в плазме биологически активного гормона роста повышен. Отсутствие эффекта гормона роста объясняется снижением уровня соматомединов (прежде всего ИФР-1), синтез которых не повышается при введении экзогенного соматотропина. Описаны семейные случаи заболевания, часто в таких семьях регистрируются кровнородственные браки. Заболевание наследуется по аутосомно-рецессивному типу, но возможны и спорадические случаи.
Помимо дефицита СТГ (или нарушения механизма его действия) выраженная задержка роста у детей может быть обусловлена дефицитом и других анаболических гормонов (тиреоидных, половых, инсулина). Так, для больных гипотиреозом, гипогонадизмом (в подростковом возрасте) и синдромом Мориака (при тяжелом течении сахарного диабета) характерны задержка роста и отставание костного возраста. Выраженная низкорослость как результат преждевременного закрытия зон роста всегда имеет место у больных с преждевременным половым созреванием любой этиологии. Высокий уровень глюкокортикоидов, как ятрогенный, так и эндогенный (болезнь, синдром Иценко—Кушинга), также приводит к нарушению процесса линейного роста. Механизм основан на непосредственном нарушении метаболизма, в том числе повышении катаболизма белков для получения энергии, уменьшении липолиза и снижении синтеза коллагена. Глюкокортикоиды также оказывают угнетающее воздействие на продукцию гормона роста, повышая содержание соматостатина и подавляя интермиттирующее высвобождение СТГ. Кроме того, они подавляют продукцию ИФР-1. Результатом всех этих процессов является тот факт, что большинство детей с избытком стероидов имеют небольшой рост. В типичных случаях у них увеличено отношение веса к длине тела и развивается ожирение. В отличие от взрослых больных с синдромом Иценко-Кушинга у детей ожирение обычно развивается пропорционально; кроме того, отличительным признаком такого заболевания является мышечная слабость.
Сложность при дифференциальной диагностике нарушений роста при этих состояниях может возникнуть лишь в том случае, когда у больного с легким вариантом первичного врожденного гипотиреоза имеют место пропорциональная задержка роста и отставание костного возраста (моносимптомный вариант заболевания) и нет других типичных симптомов врожденного гипотиреоза. Особенности гормонального профиля этих заболеваний позволяют без труда установить правильный диагноз. Повышение уровня ТТГ и нормальный на фоне стимуляции уровень СТГ (после насыщения ТГ) позволяют исключить у больного дефицит СТГ как причину задержки роста и установить диагноз первичного гипотиреоза (моносимптомный вариант).
Псевдогипопаратиреоз (синдром Олбрайта) также можно отнести к эндокриннозависимым вариантам задержки роста. При этом синдроме паращитовидные железы гистологически нормальны и способны синтезировать и секретировать паратгормон. Заболевание обусловлено наследственным дефектом рецепторных тканей, в частности почек и скелета, клетки-мишени резистентны к действию паратгормона. Для больных псевдогипопаратиреозом характерны низкорослость, возникающая в возрасте после 3—4 лет, короткая шея, короткие и широкие фаланги пальцев, брахидактилия, чаще всего 1, 4, 5-й пястных костей, а также 1-й и 5-й плюсневых; экзостозы и утолщение свода черепа, генерализованная деминерализация костей. У таких больных нередко обнаруживаются отложения кальция и метапластическое образование костной ткани под кожей. Часто отмечаются умственная отсталость, кальцификация базальных ганглиев и катаракта. Подтверждением диагноза служат гипофосфатурия и гипокальциемия, низкий уровень паратгормона в крови.
Эндокриннонезависимые варианты задержки роста. Значительно чаще у больных с задержкой роста признаки нарушения функции эндокринных желез отсутствуют, то есть у большинства детей задержка роста обусловлена неэндокринными факторами.
Тяжелые соматические заболевания, в результате которых возникают состояния длительной гипоксии (врожденные пороки сердца, анемия, заболевания легких), нарушения всасывания (целиакия, муковисцидоз), тяжелые метаболические нарушения (хронические заболевания печени и почек), а также патология костной системы (хондродистрофия, гаргоилизм и другие врожденные синдромы) часто сопровождаются выраженной задержкой роста. При данных вариантах нанизма признаки первичного нарушения функции эндокринных желез отсутствуют, костный возраст, как правило, соответствует хронологическому. На первый план выступают симптомы основного заболевания, что позволяет без труда установить причину задержки роста.
Конституциональная задержка роста — самая частая проблема, с которой сталкивается педиатр.
Конституциональная задержка роста и полового созревания — синдром позднего пубертата — характеризуется особенностями роста и развития наследственного характера, встречается примерно у 2 % детей, чаще у мальчиков. Обычно родители и (или) ближайшие родственники этих детей имеют те же особенности развития. Так, длина и масса тела при рождении не отличаются от таковых у здоровых детей. Наиболее низкие показатели темпов роста имеют место в первые годы жизни, и, следовательно, наиболее выраженная задержка роста наблюдается у детей в возрасте 3—4 лет. С 4—5-летнего возраста темпы роста восстанавливаются (5—6 см в год), однако, имея исходно низкий рост, дети остаются в школьном возрасте низкорослыми. Костный возраст несколько (в среднем на 2 года) отстает от хронологического. Этим обстоятельством можно объяснить позднее вступление в пубертат: обычно половое развитие и, следовательно, пубертатный скачок в росте запаздывают у этих детей на 2—4 года. В связи с этим подростки с синдромом позднего пубертата временно отстают в своем развитии (по показателям физического развития) от сверстников. Позднее вступление в пубертат в данном случае следует признать благоприятным фактором, так как оно позволяет пациентам с подобными особенностями конституционального развития иметь в конечном итоге нормальный рост.
Семейная низкорослость. Среди родственников детей с подобным вариантом задержки роста всегда имеются низкорослые. При рождении дети имеют нормальные показатели роста и массы тела, но темпы роста в возрасте после 3—4 лет составляют 2—4 см в год. Костный возраст этих детей обычно соответствует хронологическому, и, следовательно, вступление детей в пубертат соответствует нормальным срокам. Данное обстоятельство является причиной низкорослости взрослых пациентов с этими особенностями развития. Нельзя исключить, что причиной семейной низкорослости являются конституциональные особенности синтеза и секреции гормона роста.
Примордиальный (внутриутробный, первичный) нанизм. Особенностью данного варианта задержки роста является нарушение процессов роста с периода внутриутробной жизни. Доношенные новорожденные с этой патологией имеют недостаточную длину и массу тела. На всех этапах жизни дети с примордиальным нанизмом значительно отстают от своих сверстников в росте. Однако в отличие от детей с эндокриннозависимой задержкой роста костный возраст у этих детей соответствует хронологическому, пубертатный период, как правило, наступает в обычные сроки. Уровень СТГ соответствует нормальным показателям. Не вызывает сомнения факт, что группа детей с примордиальным нанизмом гетерогенна. В эту группу больные объединяются по одному главному признаку — нарушению процессов роста с периода внутриутробной жизни (генетические синдромы Рассела-Сильвера, Секкеля и др., внутриутробная инфекция (краснуха, сифилис, токсоплазмоз, цитомегалия), «плод алкоголика» и др.). Для больных синдромом Рассела-Сильвера характерны низкорослость, нависание лба, маленькое треугольной формы лицо, почти полное отсутствие подкожно-жировой клетчатки, укорочение и искривление 5-го пальца; во многих случаях отмечается асимметрия лица. Уровень СТГ обычно не отличается от нормы, однако в единичных случаях может встречаться недостаточность гормона роста.
Основными признаками синдрома Секкеля (карликовость с «птицеголовостью») являются дефицит длины и массы тела при рождении, микроцефалия, узкое лицо, большой клювовидный нос, редкие волосы, низко посаженные деформированные ушные раковины, ретрогнатия, аномалии скелета; характерна умственная отсталость.
Из аномалий половых хромосом к низкорослости может привести синдром Шерешвеского— Тернера. При классическом варианте синдрома (кариотип 45,ХО) рост взрослых больных не превышает 142—145 см, при мозаицизме (кариотип 45,ХО/46,ХХ) рост может быть несколько выше. При рождении дети с этим синдромом имеют нормальные показатели длины и массы тела, задержка роста начинает обращать на себя внимание с 2—3-летнего возраста. С этого времени темпы роста снижаются до 2—3 см в год. Костный возраст, как правило, до 11 — 12 лет соответствует хронологическому, в дальнейшем из-за выраженного гипогонадизма отстает от хронологического. При классическом варианте заболевания вторичные половые признаки отсутствуют, при мозаицизме могут быть выражены в разной степени. Типичные признаки кроме низкого роста: короткая шея, крыловидные кожные складки на шее, широкая грудная клетка, низкая линия роста волос, половой инфантилизм, первичная аменорея, бесплодие. Возможно поражение сердечно-сосудистой системы (коарктация аорты, дефект межжелудочковой перегородки, гипертензия), аномалии мочевой системы. В 16 % случаев полной моносомии X обнаружено снижение умственного развития. Для подтверждения диагноза необходимо исследовать половой хроматин, кариотип больного.
Другим синдромом, близким по фенотипу к синдрому Шерешевского-Тернера, является синдром Нуннан, при котором наряду с низкорослостью и крыловидными складками на шее, врожденными пороками сердца и почек обнаруживаются выраженные дисморфии лица (птоз, гипертелоризм, экзофтальм, микрогнатизм). Кариотип при этом синдроме нормальный, болеют как мальчики, так и девочки.
Нанизм возникает также и в результате поражения костной системы. Для некоторых заболеваний этой группы установлен наследственный характер, этиология других до конца не выяснена. К этой группе относятся хондродистрофия, синдром Эллиса-Ван-Кревельда, остеохондродистрофия, несовершенный остеогенез, врожденная эпифизарная точечная дисплазия, фиброзная остеодисплазия, нейрофиброматоз Реклингхаузена и некоторые формы мукоролисахаридоза.
При хондродистрофии больные отличаются карликовым непропорциональным ростом, ведущим симптомом является укорочение конечностей в основном за счет проксимальных отделов. Туловище сохраняет нормальные возрастные размеры, хотя позвоночник лишен нормальных изгибов. При рентгенологическом обследовании обнаруживаются узурированные эпифизарные хрящи, дистрофия метафизов, смещенность эпифизов.
Среди причин низкорослости следует также отметить наследственные рахитоподобные заболевания (фосфат-диабет, болезнь и синдром де Тони-Дебре-Фанкони, почечный тубулярный ацидоз — синдром Батлера-Олбрайта, проявляется в дошкольном возрасте, чаще наблюдается у девочек (70 %); синдром Лоу, встречающийся лишь у мальчиков и характеризующийся мышечной гипотонией, врожденной катарактой, метаболическим ацидозом).
СЕМИОТИКА ВЫСОКОГО РОСТА.
Обычно чрезмерный рост у детей заставляет родителей обращаться за помощью в случаях сопутствующих отклонений от нормы в виде утомляемости, частых заболеваний, нарушений полового развития и др. При их отсутствии ускоренный рост редко является причиной беспокойства.
Чаще всего, если не считать генетически высоких людей, у детей наблюдается конституциональное ускорение роста. У таких детей ускоряются созревание костей, рост и рано наступает пубертатный период. Конституционально высокие люди пропорционально сложены, у них отсутствуют признаки повышенного внутричерепного давления.
Ожирение в препубертатном возрасте может привести к ускоренному росту, однако это явление временное; дети могут быть высокими, но не достигают гигантского роста.
При открытых зонах роста избыток гормона роста у детей приводит к развитию гигантизма, а не акромегалии, как у взрослых, однако признаки акромегалии бывают и при гигантизме даже у детей и подростков, а после закрытия эпифизов акромегалия становится более выраженной.
Избыточная продукция соматотропного гормона может привести к ускоренному росту ребенка при каких-либо процессах (гидроцефалия, перенесенный энцефалит), стимулирующих активность гипоталамо-гипофизарной области. В этом случае гигантизм может развиться в любом возрасте, но наиболее часто ускорение роста начинается в дошкольном или школьном возрасте. Для этих детей характерны легкая утомляемость, пониженная сопротивляемость к инфекциям, нескладность фигуры, слабое развитие мускулатуры, при внимательном осмотре можно обнаружить акромегалоидные черты. Гипофизарный гигантизм встречается редко. Его причиной чаще всего является эозинофильная аденома, описаны случаи гигантизма и при опухоли гипоталамуса. У двух мальчиков с синдромом Олбрайта и ускоренным ростом были обнаружены функционирующие опухоли гипофиза; уровень СТГ у них был значительно повышен и не снижался при нагрузке глюкозой. В ряде случаев гигантизм и акромегалия могут начинаться как гипоталамические нарушения, которые приводят к гипертрофии и гиперплазии, а в итоге и к опухолевидному разрастанию соматотропных клеток.
Гамартомы гипоталамуса (ганглиоцитомы) способствуют развитию акромегалии, секретируя рилизинг-фактор гормона роста. Другие опухоли, особенно исходящие из поджелудочной железы, вызывают акромегалию, вырабатывая рилизинг-гормон. В большинстве случаев ускоренный рост становится очевидным в пубертатный период, но может проявиться уже в возрасте 5 лет. Конечный рост иногда достигает 250 см и более.
Часто необычно высокими для своего возраста (младший детский и начальный школьный) бывают дети с преждевременным половым развитием, но они не становятся гигантами, так как у них рано закрываются зоны роста и рост прекращается.
Высокорослых нелеченных больных тиреотоксикозом, гипогонадизмом или с синдромом Марфана (арахнодактилия) легко отличить по клиническим признакам этих заболеваний, а уровень соматотропного гормона у них нормальный.
В основе развития синдрома Кланфелтера лежит хромосомная аберрация с наличием лишней Х-хромосомы (47,XXY, 48,XXYY, 48,XXXY и др.). Больные мальчики высокого роста с непропорционально длинными конечностями, в детстве они отличаются хрупким телосложением, а в старшем возрасте у них развивается ожирение. Отличительным признаком синдрома является гипоплазия яичек и полового члена. Вторичные половые признаки развиты плохо, могут наблюдаться оволосение по женскому.типу, гинекомастия (50 % случаев). Отставание психического развития, наблюдаемое у больных, связывается с избыточной Х-хромосомой (чем их больше в кариотипе, тем выше вероятность психического отставания).
Гигантизмом, сопровождающимся повышенным уровнем СТГ, страдают некоторые больные липодистрофией, но для них характерно отсутствие подкожно-жировой клетчатки; появляется все больше данных о нарушении функции гипоталамуса при этом заболевании.
Церебральный гигантизм (синдром Сотоса) встречается значительно чаще гипофизарного и характеризуется быстрым ростом, но уровень соматотропина в сыворотке крови остается в пределах нормы, а комплекс данных свидетельствует о том, что в основе патологии лежат мозговые нарушения. В большинстве случаев рост и масса больного превышают 90-ю процентиль, возможна макрокрания. Ребенок быстро растет, и к возрасту 1 года длина его тела превышает 97-ю процентиль. Ускоренный рост продолжается в течение 4—5 лет, а затем ребенок растет с нормальной скоростью. Пубертатный период наступает в обычные сроки или несколько раньше. Характерны большие кисти и стопы с утолщенным слоем подкожно-жировой клетчатки. Голова большая, долихоцефалической формы, нижняя челюсть выступает вперед, глаза широко расставлены, больной неловок, походка его неуклюжа, координация движений нарушена. Эти признаки почти всегда сопровождаются отставанием умственного развития, варьирующим по степени, но не прогрессирующим. На рентгенограмме определяются большой череп, высокая крыша глазниц, не измененное, но несколько наклоненное кзади турецкое седло и увеличенное расстояние между глазницами. Нередко выявляются изменения на электроэнцефалограмме и расширение желудочков мозга.
Дата добавления: 2016-09-20; просмотров: 1478;