Энзимология как учение о ферментах. Простые и сложные ферменты. 6 страница
Раздел 5.1
Энзимопатии: причины, проявления, методы диагностики.
В 1908 году английский врач Арчибальд Гаррод высказал предположение, что причиной ряда заболеваний может являться отсутствие какого-либо из ключевых ферментов, участвующих в обмене веществ. Он ввёл понятие "inborn errors of metabolism" (врождённый дефект обмена веществ). В дальнейшем эта теория была подтверждена новыми данными, полученными в области молекулярной биологии и патологической биохимии.
Информация о последовательности аминокислот в полипептидной цепи белка записана в соответствующем участке молекулы ДНК в виде последовательности тринуклеотидных фрагментов - триплетов или кодонов. Каждый триплет кодирует определённую аминокислоту. Такое соответствие называется генетическим кодом. Причём некоторые аминокислоты могут быть закодированы при помощи нескольких кодонов. Существуют также специальные кодоны, являющиеся сигналами для начала синтеза полипептидной цепи и его прекращения. К настоящему времени генетический код полностью расшифрован (таблица 5.1). Он является универсальным для всех видов живых организмов.
Таблица 5.1
Триплетный код нуклеотидов мРНК для аминокислот
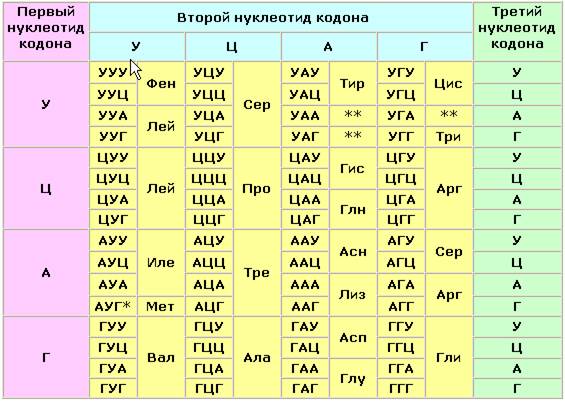
Примечания.
* - этот кодон является также сигналом для начала синтеза полипептидной цепи (инициирующий кодон);
** - эти кодоны не соответствуют ни одной из аминокислот и служат сигналом для прекращения синтеза полипептидной цепи (терминирующие кодоны).
Реализация информации, заложенной в молекуле ДНК, включает несколько этапов. Сначала в клеточном ядре в процессе транскрипции синтезируется матричная РНК (мРНК), поступающая в цитоплазму. В свою очередь, мРНК служит матрицей для трансляции - синтеза полипептидных цепей на рибосомах. Таким образом, природа молекулярных болезней определяется нарушением структуры и функции нуклеиновых кислот и контролируемых ими белков.
Мутации
Поскольку информация о структуре всех белков клетки содержится в последовательности нуклеотидов ДНК, а каждая аминокислота определяется триплетом нуклеотидов, изменение первичной структуры ДНК может в конечном счёте оказать глубокое влияние на синтезируемый белок. Подобные изменения происходят за счёт ошибок репликации ДНК, когда одно азотистое основание заменяется другим, либо в результате действия радиации или при химической модификации. Все возникшие таким образом наследуемые дефекты называются мутациями. Они могут приводить к неправильному считыванию кода и делеции (выпадению) ключевой аминокислоты, замене одной аминокислоты другой, преждевременной остановке белкового синтеза или добавлению аминокислотных последовательностей. Учитывая зависимость пространственной упаковки белка от линейной последовательности в нём аминокислот, можно полагать, что подобные дефекты способны изменить структуру белка, а значит, и его функцию. Тем не менее, многие мутации обнаруживаются только в лабораторных условиях и не оказывают вредного воздействия на функции белка. Таким образом, ключевым моментом является локализация изменений в первичной структуре. Если положение замененной аминокислоты окажется критическим для формирования третичной структуры и образования каталитического центра фермента, то мутация является серьёзной и может проявиться как заболевание.
Последствия для обмена веществ
Последствия недостаточности одного фермента в цепи реакций обмена веществ могут проявляться по-разному. Предположим, что превращение соединения A в соединение B катализирует фермент Е и что соединение C встречается на альтернативном пути превращений (рисунок 5.1):
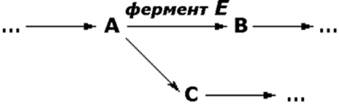
Рисунок 5.1. Схема альтернативных путей биохимических превращений.
Последствиями недостаточности фермента могут быть следующие явления:
недостаточность продукта ферментативной реакции (B). В качестве примера можно указать на снижение содержания глюкозы в крови при некоторых формах гликогенозов;
накопление вещества (A), превращение которого катализирует фермент (например, гомогентизиновая кислота при алкаптонурии). При многих лизосомных болезнях накопления, вещества, в норме подвергающиеся гидролизу в лизосомах, накапливаются в них в связи с недостаточностью одного из ферментов;
отклонение на альтернативный путь с образованием некоторых биологически активных соединений (C). К этой группе явлений относится экскреция с мочой фенилпировиноградной и фенилмолочной кислот, образующихся в организме больных фенилкетонурией в результате активации вспомогательных путей распада фенилаланина.
Если метаболическое превращение в целом регулируется по принципу обратной связи конечным продуктом, то эффекты двух последних типов аномалий будут более значительными. Так, например, при порфириях (врождённых нарушениях синтеза гема) устраняется подавляющего эффекта гема на начальные реакции синтеза, что приводит к образованию избыточных количеств промежуточных продуктов метаболического пути, которые обладают токсическим действием на клетки кожи и нервной системы.
Факторы внешней среды могут усиливать или даже полностью определять клинические проявления некоторых врождённых нарушений обмена веществ. Например, у многих пациентов с недостаточностью глюкозо-6-фосфатдегидрогеназы заболевание начинается только после приёма таких лекарственных средств, как примахин. В отсутствие контактов с лекарственными средствами такие люди производят впечатление здоровых.
Лабораторная диагностика врождённых нарушений обмена веществ
О недостаточности фермента обычно судят косвенно по повышению концентрации исходного вещества, которое в норме подвергается превращениям под действием данного фермента (например, фенилаланин при фенилкетонурии). Прямое определение активности таких ферментов проводят только в специализированных центрах, но по возможности диагноз следует подтверждать этим методом. Пренатальная (дородовая) диагностика некоторых врождённых нарушений метаболизма возможна путём иследования клеток амниотической жидкости, полученных на ранних стадиях беременности и культивируемых in vitro.
Лечение при врождённых нарушениях метаболизма
Некоторые врождённые нарушения метаболизма поддаются лечению путём доставки в организм недостающего метаболита или путём ограничения поступления в желудочно-кишечный тракт предшественников нарушенных процессов обмена веществ. Иногда могут быть удалены накапливающиеся продукты (например, железо при гемохроматозе).
Раздел 5.2
Наиболее распространённые энзимопатии.
Врождённые нарушения обмена фенилаланина и тирозина
На рисунке 5.2 представлена схема основных химических превращений фенилаланина и тирозина, а также известных в настоящее время нарушений активности ферментов, катализирующих эти реакции. Из схемы видно, что тирозин, который в норме образуется в организме из фенилаланина, является предшественником целого ряда биологически активных соединений.
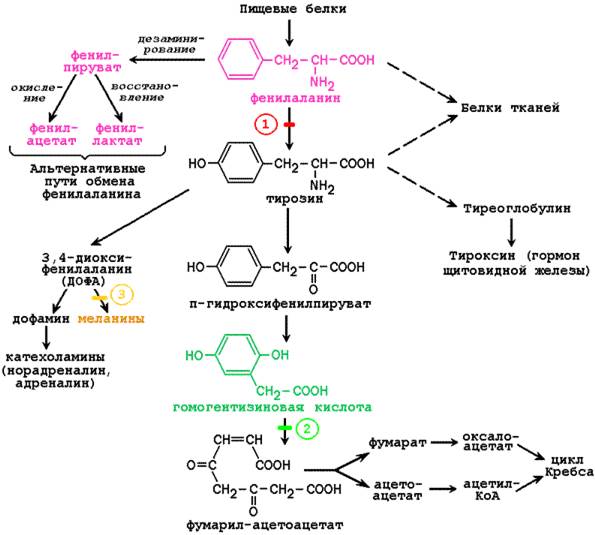
Рисунок 5.2. Обмен фенилаланина и тирозина и возможные нарушения. Цифрами показаны блокированные реакции при следующих заболеваниях: 1 - фенилкетонурия; 2 - алкаптонурия; 3 - альбинизм.
Фенилкетонурия. Это заболевание вызывается дефицитом печёночного фермента фенилаланингидроксилазы, или реже, нарушением биосинтеза его кофактора тетрагидробиоптерина. Поскольку фенилаланингидроксилаза катализирует превращение фенилаланина в тирозин, при фенилкетонурии в крови накапливается фенилаланин, который наряду с продуктами его альтернативных превращений (фенилпируват, фениллактат, фенилацетат) экскретируется с мочой. Присутствие в моче пациентов фенилпирувата (соединения, содержащего кетогруппу) нашло отражение в названии заболевания.
Избыток фенилаланина вызывает замедление транспорта тирозина и других аминокислот через клеточные мембраны. Следствием этого может быть нарушение обмена аминокислот в клетках головного мозга с последующим расстройством биосинтеза белка и нарушением синтеза нейромедиаторов. Если своевременно не выявить дефект фермента и не начать лечение, в течение первого года жизни у детей развивается умственная отсталость. В связи с этим в ряде стран широко практикуются скрининговые исследования новорождённых с целью раннего выявления случаев фенилкетонурии (определение концентрации фенилаланина в крови, взятой из прокола кожи на пятке).
Лечение заболевания заключается в том, чтобы ограничить поступление фенилаланина с пищей. С этой целью используются искусственные питательные смеси. По современным рекомендациям, длительность лечения фенилкетонурии составляет от 5 до 10 лет.
Алкаптонурия. Это заболевание обусловлено врождённой недостаточностью оксидазы гомогентизиновой кислоты. Гомогентизиновая кислота накапливается в крови, тканях и моче. Окисление и полимеризация этого вещества приводит к образованию пигмента алкаптона. Отложение алкаптона в хрящах, которые затем темнеют, называется охронозом. Превращению гомогентизиновой кислоты в алкаптон способствует щелочная среда; при алкаптонурии наиболее явным симптомом является экскреция либо чёрной мочи, либо мочи, которая темнеет по мере защелачивания при хранении.
Алкаптонурия в большинстве случаев не требует специального лечения, но в среднем возрасте и позже обычно развивается артрит.
Альбинизм. Недостаточность фермента тирозиназы в меланоцитах (пигментных клетках кожи и радужной оболочки глаз) вызывает одну из форм альбинизма и наследуется как рецессивный признак. У пациентов отсутствует пигментация кожи, волос и радужной оболочки (глаза кажутся розовыми). Отсутствие пигмента в коже сопровождается повышенной чувствительностью к солнечным лучам. Следует отметить, что биосинтез адреналина у альбиносов не нарушается, так как тирозиназа, участвующая в образовании катехоламинов, представляет собой другой фермент, контролируемый иным геном.
Галактоземия
Галактоземия - врождённое нарушение обмена веществ, обусловленное недостаточностью фермента галактозо-1-фосфатуридилтрансферазы. Для галактоземии характерна триада симптомов: увеличение размера печени, катаракта и умственная отсталость. В крови больных повышено содержание галактозы, этот моносахарид обнаруживается и в моче. Первые признаки заболевания у ребёнка (диарея, рвота, обезвоживание) выявляются очень рано, обычно через несколько дней после начала грудного вскармливания.
Источником галактозы в организме является дисахарид лактоза, содержащаяся в молоке. После расщепления лактозы в микроворсинках слизистой оболочки тонкого кишечника галактоза под действием фермента галактокиназы превращается в печени в галактозо-1-фосфат (рисунок 5.3).
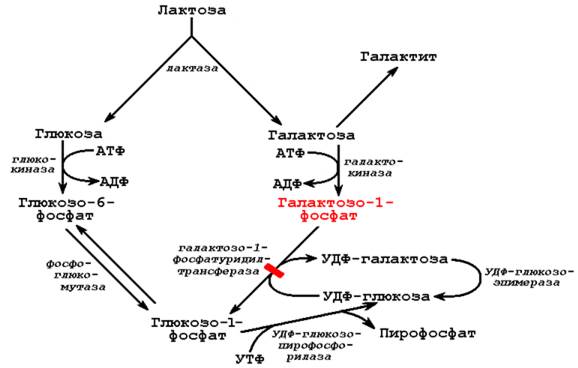
Рисунок 5.3. Обмен галактозы и основная причина галактоземии.
В нормальных условиях галактозо-1-фосфат под влиянием галактозо-1-фосфатуридилтрансферазы переходит в УДФ-галактозу. В результате угнетения этой реакции в организме накапливается галактозо-1-фосфат - метаболит с очень коротким в нормальных условиях периодом существования. В связи с этим в норме он не вызывает нарушений. Однако при накоплении галактозо-1-фосфата проявляется его мощное токсическое действие. Природа токсического влияния галактозо-1-фосфата, вероятно, объясняется структурным сходством галактозы с глюкозой. Галактозо-1-фосфат, присоединяясь к активному центру ферментов, метаболизирующих глюкозо-1-фосфат, блокирует их, что приводит к нарушению обмена глюкозы. Так, в клетках печени накопление галактозо-1-фосфата вызывает ингибирование фосфоглюкомутазы и глюкозо-6-фосфатазы - ферментов, участвующих в превращении гликогена в глюкозу, в результате чего снижается уровень глюкозы в крови. В хрусталике глаза избыток галактозы переходит в шестиатомный спирт галактит. Галактит не подвергается дальнейшим превращениям и приводит к набуханию соединительной ткани и развитию катаракты. В клетках головного мозга нарушается синтез гликолипидов вследствие недостаточного образования их предшественника УДФ-галактозы.
Если галактозу не исключить из диеты, возможны тяжёлые последствия, в том числе летальный исход.
Гликогенозы
Этот термин является общим для группы наследственных заболеваний, характеризующихся отложением в тканях аномально больших количеств полисахарида - гликогена, являющегося важным источником энергии и резервом углеводов в тканях. Врождённые нарушения содержания и структуры гликогена обусловлены дефицитом одного из ферментов, участвующих в расщеплении гликогена в печени или в скелетных мышцах (рисунок 5.4).
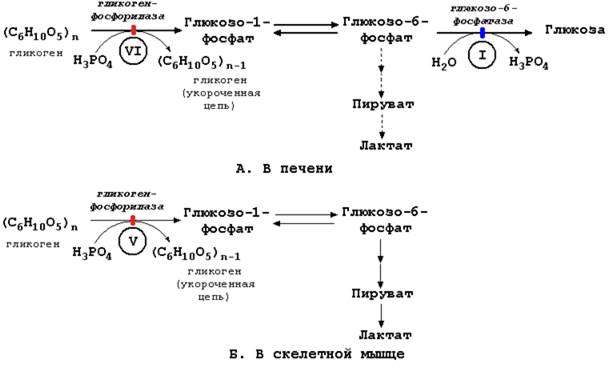
Рисунок 5.4. Расщепление гликогена в печени и скелетных мышцах и его нарушения.
Примеры:
Гликогеноз I типа (болезнь Гирке) – дефицит глюкозо-6-фосфатазы в печени. Характеризуется повышенным содержанием гликогена в печени; содержание глюкозы в крови снижено. После введения адреналина или глюкагона (гормонов, активирующих фермент гликогенфосфорилазу), уровень пирувата и лактата в крови существенно возрастает.
Гликогеноз V типа (болезнь Мак-Ардля) – дефицит фосфорилазы в скелетных мышцах. У больных развивается пониженная выносливость к физическим нагрузкам. В скелетных мышцах содержится аномально высокое количество гликогена. Тем не менее, после выполнения физической работы или после введения адреналина содержание лактата в крови не увеличивается.
Гликогеноз VI типа (болезнь Херса) – дефицит фосфорилазы в печени. Для этого заболевания характерно повышение содержания гликогена в печени, гипогликемия. После введения адреналина или глюкагона содержание лактата в крови не увеличивается (в отличие от гликогеноза I типа).
Раздел 5.3
Приобретённые нарушения ферментативной активности и их последствия.
Не во всех случаях недостаток фермента в организме связан с генетическими дефектами. Например, при воспалительных заболеваниях и опухолях желудка нарушается выделение соляной кислоты и желудочных ферментов. В результате нарушается переваривание пищи, размножаются микроорганизмы, усиливаются процессы брожения и гниения. Недостаточность поджелудочной железы, развивающаяся при хроническом алкоголизме, сопровождается снижением выработки и секреции в просвет двенадцатиперстной кишки трипсиногена, химотрипсиногена, липазы и амилазы. Это приводит к нарушению переваривания и всасывания белков, жиров и углеводов в тонком кишечнике.
Другой пример нарушения функционирования ферментов может быть связан с недостаточностью биосинтеза небелковой части сложного фермента (кофермента или простетической группы). Биосинтез ферментного белка может осуществляться нормально, но тем не менее активность фермента в тканях понижена. Как вам известно из материалов темы 1, большинство коферментов является производными витаминов. Витамины в организме человека не синтезируются и поэтому должны поступать в готовом виде с пищей. Если тот или иной витамин в пище отсутствует, то соответствующий кофермент не может быть построен и биохимические реакции, в которых он участвует, не происходят. Заболевания, обусловленные отсутствием витаминов в организме, называют авитаминозами. По существу они являются аферментозами.
Авитаминозы — полное истощение витаминных ресурсов организма, проявляющееся на фоне специфических клинических симптомов, характерных для конкретного витамина или их группы. Авитаминозы развиваются, как правило, на фоне длительного голодания.
Чаще всего приходится встречаться с гиповитаминозными состояниями. Гиповитаминозом считают снижение содержания витаминов в организме по сравнению с его нормальными потребностями. Клинически гиповитаминоз проявляется отдельными нерезко выраженными проявлениями, характерными и для этого вида авитаминоза. Неспецифическими проявлениями гиповитаминозов могут быть общие для различных видов гиповитаминозов или полигиповитаминозов симптомы: снижение аппетита, работоспособности, быстрая утомляемость и др. Причины, приводящие к развитию гиповитаминозов, многообразны, но в основном их можно разделить на 4 группы:
алиментарная недостаточность витаминов;
угнетение нормальной кишечной микрофлоры, продуцирующей ряд витаминов;
нарушение усвояемости (ассимиляции) витаминов;
повышенная потребность организма в витаминах.
Давно известна и когда-то была широко распространена болезнь «бери-бери» (сейчас ее называют полиневритом — множественным воспалением нервов, в некоторых слаборазвитых странах она и теперь встречается нередко). Причина ее— отсутствие в пище витамина В1. Этот витамин— тиамин — в соединении с фосфорной кислотой представляет собой небелковую часть фермента декарбоксилазы, с которым мы уже встречались: декарбоксилаза разрушает карбоксильную группу (СООН) некоторых органических кислот, отщепляя от нее углекислоту (СО2). Для обмена веществ в нервной системе особую роль играет декарбоксилирование пировиноградной кислоты. В отсутствие витамина В1 декарбоксилаза образоваться не может, реакция прекращается и в нервной ткани наступают нарушения, типичные для полиневрита: параличи конечностей, боли в мышцах, слабость, контрактуры.
Тяжелое заболевание — пеллагра — связано с отсутствием в пище витамина РР — никотиновой кислоты. Название «пеллагра» происходит от двух итальянских слов, которые по-русски означают шершавая кожа. При пеллагре наблюдаются воспаление кожи, нарушения деятельности кишечника и психические расстройства. Интересно отметить, что связь пеллагры с недостатком никотиновой кислоты была установлена только в 1937 г., в то время как само вещество известно химикам с 1866 г.
Сейчас мы знаем, что причина нарушений при авитаминозе РР заключается в том, что никотиновая кислота в форме никотинамида входит в состав кофермента — небелковой части большой группы окислительных ферментов — никотинамидных дегидрогеназ, о которых мы подробно рассказывали, когда обсуждали процесс биологического окисления. Мы не вдавались в тонкости их химической структуры, приводили лишь их сокращенные обозначения НАД и НАДФ и подчеркивали, что никотинамид представляет собой их главную составную часть.
Естественно, что в отсутствие никотинамида фермент не работает. Следовательно, и здесь, в случае пеллагры, авитаминоз обернулся аферментозом.
По некоторым данным, пеллагра вызывается отсутствием в пище не одной только никотиновой кислоты, но и еще одного соединения — пиридоксина, известного под названием витамина В6. Считают, что нервные расстройства, наблюдаемые при пеллагре у людей, зависят именно от отсутствия пиридоксина. На протяжении двух-трех последних десятилетий, главным образом благодаря исследованиям советского биохимика академика А. Е. Браунштейна и его школы, было показано, что производные пиридоксина в соединении с фосфорной кислотой входят в состав многих ферментов обмена аминокислот. Недостаток в пище пиридоксина — вещества, не синтезируемого в организме человека, приводит к невозможности построить нуждающиеся в нем ферменты, а это в свою очередь ведет к нарушениям обмена веществ и к характерным заболеваниям.
Упомянем еще об одном витамине. Он называется витамином В2, а по химической природе представляет собой довольно сложную циклическую структуру — рибофлавин. Авитаминоз В2 связан с тяжелыми поражениями кожи лица и глаз. Причина — недостаток фермента. Вспомним, что в цепи биологического окисления кроме никотинамидных дегидрогеназ участвует еще одна группа сходных с ними ферментов — флавиновые дегидрогеназы. Именно они не могут быть синтезированы в организме при отсутствии в пище рибофлавина, а их недостаток проявляется в форме тяжелого заболевания.
Таким образом, при самых различных авитаминозах развитие заболевания связано с отсутствием или недостатком ферментов, которые не могут быть синтезированы в организме из-за отсутствия необходимых витаминов. В большинстве случаев лечение таких заболеваний заключается в восполнении витаминных запасов организма. Профилактика витаминной недостаточности состоит в обеспечении полного соответствия между потребностями человека в витаминах и их поступлением с пищей. Наряду с полноценным витаминным составом рацион должен быть оптимален по своей энергетической ценности, содержать соответствующие количества других пищевых веществ, прежде всего незаменимых. При этом особенно важно достаточное поступление с пищей полноценного белка, дефицит которого может вести к нарушению процессов усвоения витаминов в желудочно-кишечном тракте, их транспорта в крови, внутриклеточного метаболизма и др. Обязательным требованием является сбалансированность между всеми заменимыми и незаменимыми факторами питания.
Основные проявления некоторых гипо- и авитаминозов представлены в таблице 5.2.
Таблица 5.2
Характеристика гиповитаминозов
иамин (витамин B1) бери-бери Мышечная слабость, истощение, нарушение координации движений, периферический неврит, сердечная недостаточность, в крови резко возрастает концентрация пировиноградной кислоты
рибофлавин (витамин B2) Очаговое выпадение волос, повреждение слизистой рта, изъязвление углов рта и глоссит, воспаление роговицы глаз, катаракта хрусталика
ниацин (витамин PP) пеллагра Дерматит (поражение кожи), диарея (поражение желудочно-кишечного тракта), деменция (нарушения нервной деятельности, слабоумие)
биотин (витамин H) себорея Очаговое выпадение волос, анемия, потеря аппетита и тошнота, депрессия, слабость, болезненность и слабость мышц, сухость и сероватый оттенок кожи
цианкобаламин (витамин B12) злокачественная анемия Анемия, поражение желудочно-кишечного тракта, утомляемость, онемение и другие нервные нарушения, аномалии сердечного ритма
фолиевая кислота (витамин Bc) мегалобластическая анемия Снижение количества эритроцитов и гемоглобина в крови; появление в крови и костном мозге крупных клеток – мегалобластов
аскорбиновая кислота (витамин C) цинга Кровоточивость дёсен, выпадение зубов, подкожные кровоизлияния, медленное заживление ран, потеря волос, слабость, раздражительность
Раздел 5.4
Изменения активности ферментов в тканях под действием лекарственных препаратов и ядов.
Механизмы токсического действия подавляющего большинства химических веществ в настоящее время неизвестны. В этой связи, очень многие описываемые ниже классы молекул и молекулярных комплексов, образующих организм, рассматриваются, по большей части, лишь как вероятные рецепторы (мишени) действия ядов. Рассмотрение их в этом ракурсе правомочно, поскольку в основе действия некоторых хорошо изученных токсинов лежит взаимодействие с представителями именно этих классов биомолекул.
Структурными элементами клеток, с которыми взаимодействуют токсические вещества, как правило, являются: белки; нуклеиновые кислоты; липидные элементы биомембран; селективные рецепторы эндогенных биорегуляторов (гормонов, нейромедиаторов и т.д.).
При взаимодействии ядовитых веществ с белками токсический эффект может развиваться при нарушении каждой из функций белков (транспортной, структурной, каталитической).
К числу веществ, денатурирующих белки, относятся крепкие щелочи, кислоты, окислители, ионы тяжелых металлов. В основе денатурации лежит повреждение внутримолекулярных связей, поддерживающих вторичную, третичную структуру белка. При этом наиболее часто токсические соединения взаимодействуют с СООН-, NH-, OH-, SH-группами аминокислот, образующих белки. Многочисленные токсины, связывающиеся с SH-группами, называются тиоловыми ядами. К числу тиоловых ядов прежде всего следует отнести тяжелые металлы, такие как ртуть, мышьяк, сурьма, таллий, органические соединения этих металлов (метилртуть, люизит и т.д.). Другие металлы более активно взаимодействуют с карбоксильными группами (свинец, кадмий, никель, медь, марганец, кобальт).
Особое значение в токсикологии придают действию чужеродных веществ (ксенобиотиков) на ферменты. Возможными механизмами модуляции активности ферментов химическими веществами являются:
1. Усиление каталитической активности
усиление синтеза энзимов
блокада разрушения ферментов
активация ферментов
2. Угнетение каталитической активности
угнетение синтеза ферментов
ускорение разрушения ферментов
угнетение специфической активности
3. Изменение конформации ферментов
Усиление каталитической активности ферментов
Это действие может быть вызвано поступлением в организм индукторов синтеза белков.
Физиологическими индукторами синтеза ферментов являются многие субстраты и вещества, повышающие содержание коферментов в биосредах. Некоторые гормоны выступают в качестве индукторов синтеза белка. Так, трииодтиронин у крыс с удалённой щитовидной железой существенно увеличивает содержание глюкозо-6-фосфатазы и НАДН-цитохром-с-редуктазы в микросомах печени. Стероидные гормоны - активные индукторы синтеза ферментов, например, триптофанпирролазы и др.
К числу индукторов относятся барбитураты, циклические углеводороды, полигалогенированные полициклические углеводороды и многие другие. Токсичность такого известного токсиканта, как 2,3,7,8-тетрахлорпарадибензодиоксин (диоксин, ТХДД) в настоящее время связывают именно со способностью вызывать индукцию синтеза ферментов. Среди индукторов многие - канцерогены. Например, 3,4-бенз(а)пирен, 5-метилхолантрен.
Активность фермента зависит от наличия в среде кофакторов или простетических групп. Функции кофакторов выполняют различные производные витаминов и ионы металлов. Их поступление в организм необходимо, однако передозировка сопровождается интоксикацией. Особенно опасно перенасыщение организма жирорастворимыми витаминами (А, D). Стойкое повышение содержания ионов кальция в цитоплазме клеток, отмечаемое при интоксикациях некоторыми веществами, сопровождается чрезмерной активацией ряда ферментов (см. ниже).
Существенное влияние на активность ферментов оказывают вещества, блокирующие процессы их разрушения. Все белковые молекулы в организме имеют определенное время жизни. Процессы непрерывающегося синтеза уравновешиваются столь же постоянным разрушением белка. Период полусуществования молекул ферментов колеблется в широких пределах. Например, для альдолазы мышечной ткани крыс он составляет около 20 дней, для каталазы - 1 день, для триптофанпирролазы печени - 2 часа. В процессе разрушения ферментов принимают участие протеазы и эндопептидазы. Разрушение короткоживущих белков осуществляется также энзимами аппарата Гольджи. Ингибиторами разрушения ферментов (и других белков) являются ингибиторы протеаз/пептидаз. К их числу, относятся некоторые карбамилфосфаты.
Разрушение SH-содержащих ферментов иногда начинается с окисления этих групп. Ксенобиотики с высоким восстановительным потенциалом, защищая сульфгидрильные группы, могут предотвращать разрушение ферментов. Эти эффекты также могут лежать в основе токсического процесса.
Особую роль в токсикологии играют механизмы активации лизосомальных ферментов, вызывающих, при выходе в цитоплазму, аутолиз клеток. Посредством такого механизма действуют на организм многочисленные вещества, например, иприты, СCl4, и т.д.
Угнетение каталитической активности
Снижение активности ферментов при действии токсикантов может быть следствием трех эффектов: подавления процессов синтеза апофермента и кофакторов, активации разрушения, угнетения специфической активности.
К числу наиболее распространенных кофакторов, помимо металлов, относятся железопорфирины, флавины, никотинамид-адениндинуклеотид (НАД), пиридоксальфосфат, тиаминпирофосфат и др. Отчасти эти вещества синтезируются в организме животных и человека, отчасти попадают с пищей в форме витаминов. Некоторые вещества являются конкурентами кофакторов ферментов. Так, дикумарол конкурентно препятствует утилизации печенью витамина К, необходимого для синтеза протромбина, поэтому через 24 - 96 ч после поступления вещества в организм в токсических дозах возможно развитие кровотечений угрожающих жизни.
Некоторые токсиканты нарушают образование коферментов, предшественники которых поступают в организм с пищей. Так, гидразин и его производные, взаимодействуя с пиридоксалем, содержащимся в клетках, образуют пиридоксальгидразоны, которые, в свою очередь, угнетают активность пиридоксалькиназы и блокируют тем самым синтез в организме пиридоксальфосфата. В итоге понижается активность большого числа ферментов, кофактором которых является пиридоксальфосфат (декарбоксилазы, трансаминазы и т.д.).
Дата добавления: 2016-04-02; просмотров: 843;