Високочастотне кондуктометрическое титрування
Метод заснований на вимірі електропровідності при частотах трохи МГц. Істотна відмінність високочастотного титрування від низькочастотного полягає в тому, що електроди можна розташувати на зовнішніх стінках кондуктометричної комірки. Ця відмінність важлива при роботі з агресивними середовищами (царська горілка, олеум).
Цей метод точний і дає можливість вимірювати електропровідність будь-яких середовищ.
Лекція 6. Потенціометричні методи аналізу
План лекції
1. Теоретичні основи метода. Електродні системи. Індикаторні електроди і електроди порівняння.
2. Прямий метод та потенціометричне титрування.
3.Прилади для потенціометричних вимірювань.
1. Потенціометричний аналіз належить до електрохімічних методів аналізу. Суть його полягає у вимірюванні потенціалу електрода, що занурений в досліджуваний розчин. Потенціометричний аналіз був розроблений ще наприкінці 19 ст., після того, як В.Г.Нернст вивів рівняння, яке пов'язує величину потенціалу металічного електрода з концентрацією іонів цього ж металу у розчині. Потенціометричні методи аналізу засновані на залежності потенціалу індикаторного електрода від концентрації потенціаловизначающих іонів. Застосовують різні електродні системи й різні електроди.
Електроди I родуявляють собою металеву пластину, занурену в розчин солі, що містить катіони цього металу. Формула Нернста зв'язує потенціал індикаторного електрода з концентрацією потенціаловизначающих іонів:
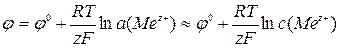 ,
, 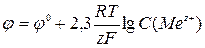 ;
; 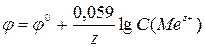 , де φ0 – стандартний електродний потенціал, обмірюваний стосовно водневого електрода при стандартних умовах; R – універсальна газова стала; Т – абсолютна температура; F – число Фарадея (96485 Кл/моль); z – число електронів, що беруть участь в елементарному акті електрохімічної реакції.
, де φ0 – стандартний електродний потенціал, обмірюваний стосовно водневого електрода при стандартних умовах; R – універсальна газова стала; Т – абсолютна температура; F – число Фарадея (96485 Кл/моль); z – число електронів, що беруть участь в елементарному акті електрохімічної реакції.
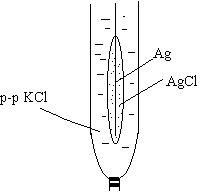 Електроди II родуявляють собою метал, покритий важко розчинною своєю сіллю й занурений у розчин солі, що містить однойменні аніони солі. До електродів другого роду відносять хлоридсрібний електрод, що представляє собою срібну пластину або дріт, покритий шаром AgCl і занурену в розчин КCl. Цей розчин сполучається з досліджуваним розчином через сольовий місток або інше пристосування, що утруднює змішання розчинів. Через азбестове волокно, по-перше, здійснюється електролітичний контакт із досліджуваним розчином, а по-друге, завдяки великому гідродинамічному опору азбесту не відбувається змішування розчину КCl з досліджуваним розчином. Формула Нернста:
Електроди II родуявляють собою метал, покритий важко розчинною своєю сіллю й занурений у розчин солі, що містить однойменні аніони солі. До електродів другого роду відносять хлоридсрібний електрод, що представляє собою срібну пластину або дріт, покритий шаром AgCl і занурену в розчин КCl. Цей розчин сполучається з досліджуваним розчином через сольовий місток або інше пристосування, що утруднює змішання розчинів. Через азбестове волокно, по-перше, здійснюється електролітичний контакт із досліджуваним розчином, а по-друге, завдяки великому гідродинамічному опору азбесту не відбувається змішування розчину КCl з досліджуваним розчином. Формула Нернста:
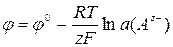 .
.
У випадку окисно-відновних систем, коли відновлена й окислена форми перебувають у розчині, електродний потенціал благородного металу (платина), зануреного в розчин цих іонів, буде дорівнює: 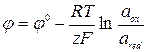 , де ox – окиснена форма, red – відновлена форма.
, де ox – окиснена форма, red – відновлена форма.
Потенціометричні комірки являють собою електродну систему з електрода порівняння, потенціал якого не залежить від природи розчину, і індикаторного електрода, потенціал якого чутливий до природи яких-небудь іонів або розчинених речовин.
Як електроди порівняння використовують 2 типи електродів II роду: хлоридсрібний (див. вище) і каломельний електрод. Каломельний електрод являє собою шар ртуті, покритий шаром каломелі Hg2Cl2, над яким перебуває розчин КCl. Контакт із зовнішнім середовищем здійснюється платиновим дротом. Розчин КCl контактує з досліджуваним через азбестове волокно. Як індикаторні електродизастосовують при дослідженні окисно-відновних систем – платинові, золоті електроди, електроди з вуглецевих матеріалів (скло вуглець, вуглецева тканина). При вимірі рН і деяких лужних металів (натрій, кальцій) використовують мембранні електроди. Вимоги до індикаторних електродів можуть бути різними в залежності від того, де вони використовуються – в абсолютній потенціометрії чи в потенціометричному титруванні. В обох випадках індикаторні електроди повинні бути оборотними, тобто їхній потенціал повинен змінюватися із зміною концентрації іонів металу в розчині відповідно до рівняння Нернста. Індикаторні електроди повинні бути хімічно стійкими по відношенню до речовин, які містяться у розчині.
pХ = –lg c(Х+); де Х – природа речовини. Наприклад, pН = –lg c(H+).
В аналітичній практиці використовують різноманітні індикаторні електроди в залежності від складу розчину – найчастіше водневі, хінгідронні та скляні.
Водневий електрод – платинова пластинка, що вкрита електролітичною губчастою платиною, яка насичена воднем. Під час занурення такого електрода у розчин з певною концентрацією гідроген-іонів між воднем, адсорбованим губчастою платиною, і іонами Гідрогену у розчині встановлюється рівновага, а потенціал електрода визначається рівнянням: Еx = E0 + 0,058 lg[H+], де Е0 - потенціал нормального водневого електрода.
Нормальним, або стандартним, водневим електродом називається водневий електрод, у якому платиновий провідник насичений воднем під тиском 101 кПа, занурений у розчин з концентрацією іонів Гідрогену 1 моль/л за температури 25°С. Нормальний потенціал водневого електрода прийнято вважати рівним нулю, тому Еx = 0,058 lg [H+]. Звідси рН = Ex/0,058.
Водневим електродом можна вимірювати концентрацію іонів Гідрогену у широких межах pH. Недоліки в роботі водневого електрода: потрібно джерело водню, який необхідно старанно очищати, підтримувати його тиск, не можна використовувати у розчинах, що містять сильні окисники або відновники.
Хінгідронний електрод – скляна посудина з платиновим електродом, зануреним у насичений розчин хінгідрону (малорозчинна молекулярна сполука хінону, яка у розчині розпадається на хінон і гідрохінон):

При вмиканні хінгідронного електрода в коло з будь-яким електродом порівняння, наприклад каломельним, на ньому відбувається реакція:

Потенціал електрода визначається рівнянням:
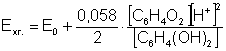
Вимірюючи потенціал хінгідронного електрода, можна легко обчислити рН або концентрацію іонів Гідрогену розчину. Величина нормального хінгідронного електрода Е0 дорівнює 0,704 В.
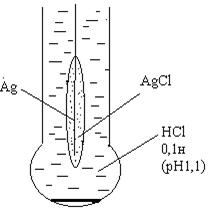 Скляний електрод – трубка з електропровідного скла, що закінчується внизу кулькою з дуже тонкою стінкою – мембраною. Зовнішня частина мембрани контактує з досліджуваним розчином. Електрод заповнений буферним розчином (0,1 н розчин НСl із рН = 1,1). Контакт із НСl здійснюють за допомогою хлоридсрібного електрода. Потенціал скляного електрода:
Скляний електрод – трубка з електропровідного скла, що закінчується внизу кулькою з дуже тонкою стінкою – мембраною. Зовнішня частина мембрани контактує з досліджуваним розчином. Електрод заповнений буферним розчином (0,1 н розчин НСl із рН = 1,1). Контакт із НСl здійснюють за допомогою хлоридсрібного електрода. Потенціал скляного електрода:
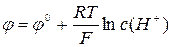 або
або 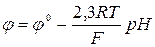 .
.
Потенціал на мембрані реалізується в результаті рівноважного процесу: переходу іонів Li+ у розчин, а іонів Гідрогену Н+ у скло. Li+стік + H+розчин↔ Li+розчин+ H+стекло. Потенціал скляного електрода перебуває у певній залежності від концентрації іонів Гідрогену, що дає змогу використовувати його для вимірювання рН.
Аналогічно поводяться електроди зі скла, чутливі до іонів Na+, Ca2+, Li+ і ін. Формула Нернста: 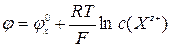 або
або 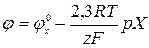 , Х – іон, до якого чутлива мембрана. Відомо багато іон-селективних електродів, чутливих до різних катіонів і аніонів.Недоліком цих електродних систем є одночасна чутливість до інших іонів.
, Х – іон, до якого чутлива мембрана. Відомо багато іон-селективних електродів, чутливих до різних катіонів і аніонів.Недоліком цих електродних систем є одночасна чутливість до інших іонів.
:
 2. Прямий метод в потенціометрії полягає в безпосередньому вимірюванні потенціалу або рН досліджуваного розчину. Оскільки потенціал є лінійною функцією від концентрації, будують калібрувальний графік залежності в координатах (φ – C), (φ – lgс) або (φ – pХ). Готують стандартні розчини з свідомо меншою й більшою концентраціями. Таке калібрування можна здійснювати, використовуючи органи підстроювання потенціометра ("калібрування" і "крутизна").
2. Прямий метод в потенціометрії полягає в безпосередньому вимірюванні потенціалу або рН досліджуваного розчину. Оскільки потенціал є лінійною функцією від концентрації, будують калібрувальний графік залежності в координатах (φ – C), (φ – lgс) або (φ – pХ). Готують стандартні розчини з свідомо меншою й більшою концентраціями. Таке калібрування можна здійснювати, використовуючи органи підстроювання потенціометра ("калібрування" і "крутизна").
В потенціометричному аналізі використовують не абсолютні значення електродних потенціалів, а зміну потенціалів в ході аналізу, наприклад при титруванні. Потенціометричне титрування – титриметричний метод аналізу, в якому точку еквівалентності визначають не за допомогою кольорових індикаторів, а за характером зміни потенціалу індикаторного електрода,який залежить від концентрації тих іонів, що визначаються.Безпосередній розрахунок за значенням електродного потенціалу застосовується для визначення концентрації іонів Гідрогену.
Потенціометричне титрування застосовують головним чином для визначення досить великих кількостей речовини. Порівняно з іншими методами має переваги: дає змогу титрувати забарвлені розчини (кольорові індикатори не придатні, як і колориметричний метод); легко визначати кілька речовин у суміші без попереднього їх розділення. На кривій титрування у цьому разі з'являється кілька стрибків потенціалу, за якими знаходять кількість робочого розчину, витраченого на взаємодію з кожним компонентом складного розчину.
У методах кислотно-основного титрування необхідно виміряти концентрацію Гідроген-іонів розчину. Якщо при титруванні в результаті реакції осадження, нейтралізації, окиснення-відновлення, комплексоутворення концентрація іонів зменшується, то зміниться і потенціал. Найбільша зміна потенціалу відбудеться в момент повного зв'язування іонів, які визначають потенціал, тобто в точці еквівалентності. Електрохімічний потенціал слугує в даному випадку індикатором в процесі титрування.
Реакції, які використовуються при потенціометричному титруванні, повинні бути практично необоротними, проходити з великою швидкістю і тільки в одному напрямі, а в точці еквівалентності повинна відбуватись зміна потенціалу індикаторного електрода (стрибок потенціалу).
Спосіб потенціометричного титрування нагадує способи роботи при класичному об'ємному аналізі. Різниця в тому, що в аналізуючий розчин занурюють електродну пару і після додавання кожної порції титранта вимірюють електродний потенціал. Необхідно інтенсивно перемішувати розчин, оскільки концентрація іонів в шарі розчину, який знаходиться біля електродів, і концентрація іонів у всьому розчині повинні бути однаковими. При титруванні склад розчину змінюється, і відповідно з новим складом розчину встановлюється новий урівноважений електродний потенціал. Це відбувається швидко, але не миттєво, і після додавання кожної порції робочого розчину потрібно витримати його впродовж деякого часу, помішуючи, після чого почати вимірювання електродного потенціалу. Результати титрування вносять до таблиці і будують криву потенціометричного титрування. На осі абсцис відкладають об'єм робочого розчину, який додається (в мілілітрах), на осі ординат – електродний потенціал (в мілівольтах) або рН. В точці еквівалентності плавний характер кривої змінюється. В залежності від значення стрибка потенціалу на кривій з'являється нерівність (при невеликому стрибку), або ступінь. Відрізок осі абсцис від нуля до точки, який означає стрибок потенціалу, відповідає об'єму робочого розчину, який витрачений на титрування до точки еквівалентності.
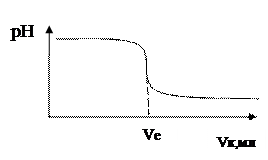 | |||
 | |||
а) б)
Рис. 14 Криві потенціометричного титрування: а) сильної кислоти лугом, б) лугу розчином кислоти.
Для більш важких аналізів будують диференціальну криву потенціометричного титрування. На осі абсцис відкладають об'єм робочого розчину (в мілілітрах), на осі ординат – різницю потенціалу, відповідно 1 мл доданого титранту. На початку титрування, навіть при додаванні великої порції робочого розчину потенціал несуттєво змінюється. По мірі приближения до точки еквівалентності кожна порція робочого розчину викликає все більшу різницю потенціалу.
 Максимальна зміна потенціалу відбувається в точці еквівалентності. Якщо по осі ординат відкладати відношення ΔE/ΔV (де ΔV – об'єм чергової порції титранта; ΔЕ – зміна потенціалу, викликана додаванням цієї порції титранта), то максимум цієї кривої буде відповідати точці еквівалентності..
Максимальна зміна потенціалу відбувається в точці еквівалентності. Якщо по осі ординат відкладати відношення ΔE/ΔV (де ΔV – об'єм чергової порції титранта; ΔЕ – зміна потенціалу, викликана додаванням цієї порції титранта), то максимум цієї кривої буде відповідати точці еквівалентності..
Крива потенціометричного титрування необхідна для визначення точки еквівалентності та використання робочого розчину. У більшості технічних аналізах, де в точці еквівалентності потенціал різко змінюється, використання робочого розчину можна визначити, не будуючи графіка. Розрахунки ведуть за формулою об'ємного аналізу: Nx = VN/Vx, де Nx – концентрація аналізуючого розчину, г-екв/л; Vх – об'єм робочого розчину, використаний на титрування до точки еквівалентності, мл; N – концентрація робочого розчину, г-екв/л.
Потенціометричне титрування в реакціях нейтралізації. Використовують звичайний електрод, потенціал якого залежить від складу в розчині іонів Гідрогену. Електрод порівняння – насичений хлоридсрібний або каломельний.
При титруванні кислот необхідно розмежовувати два випадки:
1) титрування розчину сильної кислоти розчином сильної основи;
2) титрування розчину слабкої кислоти розчином сильної основи.
В першому випадку зміна концентрації іонів водню відбувається винятково за рахунок реакції H3O+ + OH- = 2H2O. При титруванні сильної кислоти сильною основою еквівалентну точку вимірюють за допомогою точки перегину кривої. При титруванні слабких кислот стрибок потенціалу в точці еквівалентності виражений менш різко, що іноді ускладнює точне вимірювання точки перегину, тобто і кінцевого моменту титрування. При нейтралізації слабкої кислоти сильною основою в точці еквівалентності розчин буде не нейтральним, а основним, внаслідок гідролізу утвореної солі.
При титруванні суміші сильної і слабкої кислоти вдається потенціометричне встановити склад кожної з кислот окремо, не роблячи попереднього їх відокремлення. Спочатку відбувається нейтралізація сильної кислоти. Отримують два стрибки потенціалу індикаторного електрода, а потім і дві точки перегину на кривій титрування. При титруванні багатоосновних кислот сильною основою на кривій титрування отримуємо також декілька точок перегину, наявність яких пояснюється тим, що багатоосновні кислоти фактично дисоціюють в декілька стадій, причому продукти дисоціації ведуть себе подібно розчину рівних кількостей кислот. При титруванні багатоосновних кислот появу декількох точок перегину можна чекати тоді, коли відношення між константами дисоціації окремих стадій дисоціації будуть 104.
Потенціометричне титрування в реакціях осадження. При титруванні хлорид-іонів розчином аргентум нітрату індикаторним електродом слугує срібний електрод, який являє собою пластинку з листового срібла або платинову пластинку, електролітично покриту сріблом. Електрод порівняння –насичений хлоридсрібний електрод. В ході титрування в розчині утворюються молекули аргентум хлориду: NaCl + AgNO3 = AgCl + NaNO3.
Розчинність аргентум хлориду дуже мала і AgCl випадає в осад. В розчині залишається невелика кількість іонів Ag+. Концентрацію іонів Ag+ в розчині вирахувати неважко. Осадження аргентум хлориду починається тоді, коли добуток концентрації іонів Ag+ і Cl–-іонів в розчині перевищує значення добутку розчинності AgCl. В ході титрування концентрація Cl–-іонів зменшується і відповідно збільшується концентрація іонів Ag+. Але так як розчинність AgCl дуже мала, то навіть в точці еквівалентності (зв'язані всі Cl–-іони) концентрація іонів Ag+ в розчині не може перевищувати 1,3·10-5 г-іон/л ( 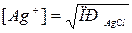 ). Коли всі Cl–-іони опиняться зв'язаними в результаті додавання наступної порції розчину AgCl, концентрація іонів Ag+ в розчині різко збільшиться. Оскільки іонами, які визначають потенціал електрода, в даному випадку являються іони Ag+, в цей момент потенціал різко змінюється (досягнення точки еквівалентності). Відкладаючи на графіку на осі абсцис об'єм доданого розчину аргентум нітрату, а на осі ординат – потенціал, отримаємо криву потенціометричного титрування. Титрування проводять двічі. Спочатку визначають приблизну витрату аргентум нітрату, додаючи його порціями по 1 мл. При повторному титруванні додають спочатку великими порціями, а по мірі приближения до точки еквівалентності по 0,1 мл. При потенціометричному титруванні за допомогою способа осадження різкий стрибок потенціалу в точці еквівалентності відбувається в тому випадку, коли в ході титрування утворюється речовина з дуже малою розчинністю. Тоді перша надлишкова крапля титранта призводить до різкої зміни електродного потенціалу. Потенціометричний метод дає можливість кількісно визначити хлориди та йодиди при взаємній їх присутності в розчині. Розчинність AgI (2,8·10-6 г/л) значно нижча, ніж розчинність AgCl (1,8·10-3 г/л). Тому при титруванні розчину, який вміщує суміш натрій йодиду та хлориду, розчином аргентум нітрату спочатку будуть осаджатися I–-іони. Після осадження всіх I–-іонів наступна крапля розчину аргентум нітрату призведе до стрибка потенціалу. Потім почнеться осадження Cl–-іонів і електродний потенціал буде визначатись концентрацією іонів Ag+. Другий стрибок потенціалу відбудеться в момент осадження Cl–-іонів. За витратою титранта на титрування до першого і до другого стрибка вираховують вміст хлориду і йодиду в аналізуючому розчині.
). Коли всі Cl–-іони опиняться зв'язаними в результаті додавання наступної порції розчину AgCl, концентрація іонів Ag+ в розчині різко збільшиться. Оскільки іонами, які визначають потенціал електрода, в даному випадку являються іони Ag+, в цей момент потенціал різко змінюється (досягнення точки еквівалентності). Відкладаючи на графіку на осі абсцис об'єм доданого розчину аргентум нітрату, а на осі ординат – потенціал, отримаємо криву потенціометричного титрування. Титрування проводять двічі. Спочатку визначають приблизну витрату аргентум нітрату, додаючи його порціями по 1 мл. При повторному титруванні додають спочатку великими порціями, а по мірі приближения до точки еквівалентності по 0,1 мл. При потенціометричному титруванні за допомогою способа осадження різкий стрибок потенціалу в точці еквівалентності відбувається в тому випадку, коли в ході титрування утворюється речовина з дуже малою розчинністю. Тоді перша надлишкова крапля титранта призводить до різкої зміни електродного потенціалу. Потенціометричний метод дає можливість кількісно визначити хлориди та йодиди при взаємній їх присутності в розчині. Розчинність AgI (2,8·10-6 г/л) значно нижча, ніж розчинність AgCl (1,8·10-3 г/л). Тому при титруванні розчину, який вміщує суміш натрій йодиду та хлориду, розчином аргентум нітрату спочатку будуть осаджатися I–-іони. Після осадження всіх I–-іонів наступна крапля розчину аргентум нітрату призведе до стрибка потенціалу. Потім почнеться осадження Cl–-іонів і електродний потенціал буде визначатись концентрацією іонів Ag+. Другий стрибок потенціалу відбудеться в момент осадження Cl–-іонів. За витратою титранта на титрування до першого і до другого стрибка вираховують вміст хлориду і йодиду в аналізуючому розчині.
Потенціометричне титрування в окисно-відновних реакціях.
В потенціометричному аналізі велике значення мають процеси, в яких беруть участь інертні електроди. Якщо занурити платиновий електрод в розчин, який містить, наприклад, іони дво- і тривалентного Феруму, що знаходяться у визначеній рівновазі: Fe2+ ⇄ Fe3+ + e, на електроді виникає потенціал, значення якого залежить від співвідношення концентрацій Fe2+- і Fe3+-іонів: Е = 0,77 + 0,0581 lg(Fe3+/Fe2+), де 0,77 – нормальний окисно-відновний потенціал системи Fe2+ ⇄ Fe3+ + e. При зміні відношення (Fe3+/Fe2+) зміниться і значення потенціалу. При цьому іони Платини в розчин не переходять, а виникнення потенціалу зв'язано з переходом електронів: платиновий електрод приймає електрони від Fe2+-іонів і передає їх Fe3+-іонам. На цьому явищі базується застосування платинового електрода при потенціометричному титруванні розчині залізного купоросу або солі Мора розчином окисника, наприклад калій перманганатом. Реакція проходить в кислому середовищі:
10FeSO4 + 2KMnO4 + 8H2SO4 = 5Fe2(SO4)3 + 2MnSO4 + K2SO4 + 8H2O
Іонне рівняння реакції має вигляд: MnO4- + 5Fe2+ + 8H+ = Mn2+ + 5Fe3+ + 4H2O
По мірі додавання до розчину залізного купоросу розчину калій перманганату вміст Fe2+ зменшується, а вміст Fe3+ збільшується. Відповідно зростає і електронний потенціал. В точці еквівалентності Ферум практично знаходиться в формі Fe3+, а Марганець – в формі Mn2+. Додавання до розчину першої краплі надлишку розчину калій перманганату призводить до виникнення нової окисно-відновної системи MnO4–/Mn2+. Значення електродного потенціалу різко змінюється. Стрибок потенціалу в точці еквівалентності тим більший, чим більша різниця потенціалів окисно-відновних процесів. При титруванні розчину залізного купоросу розчином КМnO4 стрибок потенціалу буде дуже різким, так як нормальний потенціал системи дорівнює Fe3+/Fe2+ 0,77 В, а нормальний потенціал системи MnO4–/Mn2+ –1,52 В. Чим вища початкова концентрація, тим більший стрибок в точці еквівалентності.
Переваги потенціометричного титрування:можливість робити кількісні вимірювання в забарвлених розчинах і в розчинах, які мають осад; можливість встановлювати безпосередньо і кількісно один або декілька з присутніх компонентів розчину; успішне використання багатьох реакцій нейтралізації, осадження, комплексоутворення; висока чутливість методу, який іноді перевершує чутливість звичайного об'ємного метода титрування.
Недоліки потенціометричного титрування:використання порівняно складної апаратури; необхідність розрахунків на бюретці і на вимірювальному пристрої; нестійкість в деяких випадках потенціалу індикаторного електрода або ж дуже повільне встановлення його граничного значення.
3. Для потенциометрических вимірів застосовують прилади, що вимірюють напругу постійного струму від 0 до 3 В. Такі прилади повинні відбирати від  гальванічного осередку винятково малі струми, тому звичайні вольтметри для цього методу непридатні. Використовують електронні вольтметри з більшим вхідним опором 109-1012 Ом. Вони відбирають від комірки дуже маленькі струми (109-1012 А). Звичайно ці прилади мають лампові або електронні підсилювачі й звуться потенціометри. Промисловість випускає високоомні потенціометри постійного струму ППТВ, Р-307 та інші, лампові потенціометри ЛП-5, ЛП-58, ЛПУ-01. рН-метр ЛПУ-1 використовується головним чином для вимірювання рН розчинів із скляним електродом.
гальванічного осередку винятково малі струми, тому звичайні вольтметри для цього методу непридатні. Використовують електронні вольтметри з більшим вхідним опором 109-1012 Ом. Вони відбирають від комірки дуже маленькі струми (109-1012 А). Звичайно ці прилади мають лампові або електронні підсилювачі й звуться потенціометри. Промисловість випускає високоомні потенціометри постійного струму ППТВ, Р-307 та інші, лампові потенціометри ЛП-5, ЛП-58, ЛПУ-01. рН-метр ЛПУ-1 використовується головним чином для вимірювання рН розчинів із скляним електродом.
Система з двох електродів, розташованих в дослідному розчині, являють собою гальванічний елемент, ЕРС котрого дорівнює потенціалу, що характеризує склад розчину.
Вимірювання електродного потенціалу або різниці потенціалів - непроста задача. Звичайний пристрій, який вимірює напругу, - вольтметр непридатний до вимірювання електродних потенціалів, так як на цьому пристрої відбувається втрата напруги, що призводить до значних помилок при вимірюванні. Для вимірювання електродного потенціалу частіше всього використовують компенсаційну схему (малюнок 1). Назустріч ЕРС, яка виникає в колі електродів 4, напрямляють через реохорд 2, ЕРС джерела постійного струму 1. За допомогою сковзького контакту змінюють різницю потенціалів до досягнення повної компенсації. В момент компенсації, тобто рівності ЕРС електродів та сухого елемента 1 гальванометр 3 покаже відсутність струму в колі. За положенням бігунця реохорда визначають ЕРС електродної пари, тобто електродний потенціал. В сучасних лабораторних рН-метрах, які використовуються для технічного аналізу, використовуються барабанні реохорди зі сковзьким контактом. Оскільки січна дроту реохорда по всій довжині однакова, то опір її також однаковий по всій довжині і спад напруги на кожній ділянці пропорційний довжині цієї ділянки. Якщо, наприклад, на реохорд від сухого елемента подається напруга 1,3 В, а реохорд розділений по довжині на 130 ділянок, то спад напруги на кожній ділянці дорівнюватиме 10 мВ. Відповідно шкалу реохорда також ділять на 130 ділянок. Якщо при компенсації бігунець реохорда знаходиться на дільниці 20, електродний потенціал дорівнює 200 мВ. При роботі по компенсаційній схемі в вимірювальних приладах не відбувається втрата струму і виключається можливість помилок, зв'язаних з опором системи або поляризацією.
Конструкція приладу основана на описаній вище компенсаційній схемі вимірювання. Гальванометр потрібний для визначення моменту компенсації, тобто моменту, коли в схемі немає струму. Струм, який виникає в електродному колі, дуже малий, і його посилюють за рахунок лампового підсилювача. Ламповий підсилювач – мостова схема, в якій ввімкнуті електронні лампи. В діагональ моста ввімкнутий нуль-гальванометр. Після ввімкнення підсилювача і підігріву електронних ламп налагоджують підсилювач. Для цього за допомогою спеціального перемінного опору вирівнюють анодні точки обох електронних ламп. Якщо стрілка нуль-гальванометра займає нульове положення, підсилювач налагоджений.
Потенціометрична схема слугує для вимірювання ЕРС електродної пари, попередньо підсиленої ламповим підсилювачем. Для точного виміру ЕРС електродної пари потрібно подавати на вимірюючу схему постійний струм з визначеною напругою. В лабораторних рН-метрах джерелом постійної напруги слугує постійний елемент, наприклад батарея З-СЛ-30. Оскільки її ЕРС змінюється з часом, в схему підключають нормальний гальванічний елемент Вестона. Його обрали в якості еталону тому, що має постійну ЕРС (1,0183 В при 20°С). Цей елемент підключають в схему таким чином, щоб можна було перевірити по ньому ЕРС сухого елемента. Для цього на реохорд подають ЕРС нормального елемента, підключають йому назустріч ЕРС сухого елемента і за допомогою спеціального перемінного опору зменшують ЕРС, яка подається від сухого елемента, до момента компенсації – відсутність струму в колі. Компенсація означає, що ЕРС, яка поступає від сухого елемента, дорівнює ЕРС нормального елемента, тобто 1,0183 В при 20°С. Описану операцію називають налагодженням вимірюючої схеми за допомогою нормального елемента.
Коли пристрій готовий до вимірювання, до вимірюючої схеми підключають ЕРС електродної пари. При цьому рівновага мостової схеми порушується і стрілка нуль-гальванометра відхиляється. Для компенсації відхилення ЕРС електродів протиставляють ЕРС сухого елемента. Держак реохорда обертають до тих пір, поки стрілка нуль-гальванометра знову не займе нульового положення, тобто поки ЕРС електродної пари не буде компенсована ЕРС на реохорді. За допомогою пристрою відліку реохорда відраховують ЕРС електродної пари. За допомогою потенціометра в лабораторіях виконують аналізи двох видів: знаходження концентрації іонів Гідрогену в розчині; знаходження концентрацій різних речовин за рахунок потенціометричного титрування.
Дата добавления: 2016-03-20; просмотров: 1591;