Патогенез сахарного диабета I типа
Ко времени проявления инсулинзависимого сахарного диабета большинство р-клеток поджелудочной железы уже разрушено. Деструктивный процесс почти наверняка имеет аутоиммунную природу. Патогенетическая последовательность приведена в табл. 327-2. Во-первых, должна прослеживаться генетическая предрасположенность к заболеванию. Во-вторых, у генетически предрасположенных людей причиной диабета служат факторы внешней среды. Считают, что часто это вирусные инфекции. Лучшим доказательством того, что необходим внешний толчок, послужили исследования, проведенные на монозиготных близнецах, показатель конкордантности по диабету среди которых не превышает 50%. Если бы диабет был чисто генетическим заболеванием, следовало бы ожидать, что этот показатель составит 100%. Третьим этапом в развитии заболевания является воспалительная реакция в поджелудочной железе, называемая «инсулитом»: островки инфильтрируются активированными Т-лимфоцитами. Четвертый этап заключается атакой альтерации или трансформации поверхности b-клеток, что они перестают восприниматься как «снос» и приобретают для иммунной системы свойства чужеродного, или «не своего». Пяты и этап сводится к развитию иммунной реакции. Поскольку островки воспринимаются теперь как «не свое», появляются цитотоксические антитела, которые действуют вместе с клеточно-опосредованными иммунными механизмами. Окончательный результат — разрушение b-клеток и появление клиники диабета.
Следовательно, схему патогенеза можно представить следующим образом: генетическая предрасположенность ® инсулит, вызываемый факторами окружающей среды, ® превращение b-клеток из «своего» в «не свое» ® активация иммунной системы ® разрушение b-клеток ® сахарный диабет.
Таблица 327-2. Патогенез сахарного диабета I типа
| Этап | Явление | Агент или реакция |
| Первый | Генетическая предрасположенность | HLA-DR3, DR4 (рецепторы Т-клеток?) |
| Второй | Действие факторов окружающей среды | Вирус (?) |
| Третий | Инсулит | Инфильтрация активированными Т-лимфоцитами |
| Четвёртый | Активация аутоиммуности | Переход «своего» в «не свое» |
| Пятый | Иммунная атака на бета-клетки | Антитела к островковым клеткам, клеточно-опосредованная иммунная реакция |
| Шестой | Сахарный диабет | Разрушение более 90% b-клеток (a-клетки сохранены) |
Генетика. Хотя инсулинзависимый диабет чаще встречается в определенных семьях, механизмы его наследования трудно описать в понятиях теории Менделя. Сообщалось об аутосомно-доминантной, рецессивной и смешанной формах наследования, но ни одна из них не доказана. Генетическая предрасположенность играет, по-видимому, пермиссивную, а не решающую роль.
Анализ родословных обнаруживает низкую частоту прямого вертикального наследования. По данным одного исследования, проведенного в 35 семьях, в каждой из которых имелся ребенок с классическим инсулинзависимым диабетом, только у четверых больных родители страдали диабетом и еще в двух случаях диабет был у бабушек или дедушек. Из 99 сиблингов больных диабетом детей только у 6 имелся явный диабет. Вероятность возникновения диабета I типа у детей, когда это заболевание регистрируется у другого члена семьи первой степени родства, составляет всего 5—10%. Наличие инсулиннезависимого диабета у родителей увеличивает возможность появления инсулинзависимого диабета у потомства. Отражает ли одновременное присутствие ИЗСД и ИНЗСД в одной и той же семье один генетический дефект (т. е. ИНЗСД на самом деле является ИНЗСД I типа) или в одной и той же семье случайно сочетаются два генетических дефекта, причем каждый из них, по-видимому, влияет на экспрессию другого, остается неясным. Низкие показатели передачи ИЗСД затрудняют понимание ее механизмов при семейных исследованиях, но должны ободрять больных диабетом лиц, желающих завести ребенка.
Один из генов восприимчивости к ИЗСД расположен, очевидно, на 6-й хромосоме, поскольку имеется выраженная связь между диабетом и определенными антигенами лейкоцитов человека (HLA), которые кодируются генами главного комплекса гистосовместимости, локализованными на этой хромосоме (см. гл. 63). Многие исследователи идентифицировали четыре локуса, называемые литерами А, В, С и D, с аллелями каждого сайта. Главные аллели, с которыми связан повышенный риск ИЗСД, —это HLA-DR3, HLA-Dw3, HLA-DR4, HLA-Dw4, HLA-B8 и HLA-B15. Особое значение придают локусу D, а локусы В и А вовлекаются за счет неслучайной связи c D (неравновесное сцепление). По сравнению с общей популяцией риск, обусловленный присутствием DR3 или DR4, возрастает в 4—10 раз. Если же сравнивать не с контрольной популяцией, а с группой лиц, не имеющих предрасполагающего антигена, относительный риск увеличивается в 30 раз. Однако у многих лиц, несущих аллели «высокого риска», диабет никогда не развивается.
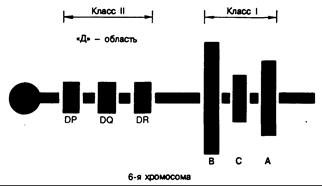
Рис. 327-1. Схематичное изображение главного комплекса гистосовместимости на 6-й хромосоме. (Любезно предоставлено д-ром J. Harold Helderman.)
Можно предположить, что дальнейшее изучение генов в D-области поможет более точно определить риск, т. е. обнаружить конкретные варианты антигена HLA-DR или HLA-DQ, не идентифицируемые при обычном скрининге, которые теснее ассоциированы с диабетом, чем простое присутствие антигена. Например, не все HLA-DR4 обусловливают повышенный риск диабета, а только некоторые их варианты. Следует подчеркнуть также, что диабет может возникнуть и при отсутствии тех HLA-детерминант, которые идентифицированы в качестве маркеров высокого риска при популяционных исследованиях. Антигены В7 и DR2 (Dw2) называют «защитными», поскольку среди больных диабетом они выявляются с меньшей частотой, чем в общей популяции. Однако на самом деле они могут быть не защитными, а аллелями «низкого риска», так как их присутствие находится в обратной зависимости от присутствия DR3/DR4. Иными словами, если присутствуют DR2, Dw2, то аллели высокого риска должны отсутствовать.
В настоящее время D-область подразделяют на участки DP, DQ и DR (рис. 327-1). (DP раньше обозначали SB, a DQ — DC.) Ассоциированный с HLA ген восприимчивости может быть теснее связан с участком DQ, чем DR. Если это так, то ассоциация с DR3 или DR4 обусловлена неравновесным сцеплением. Многие исследователи считают, что для развития диабета нужен второй ген восприимчивости, который мог бы кодировать дефект Т-клеточного рецептора.
Следует остановиться на функции тех молекул клеточной поверхности, которые кодируются генами области HLA. Антигены, кодируемые участками А, В и С, называются молекулами I класса. Они присутствуют на ядерных клетках, и их функция заключается прежде всего в защите от инфекции, особенно вирусной. Антигены D-области называются молекулами II класса. Они функционируют в сфере регуляторной (хелпер/супрессор) Т-клеточной системы и реакции на аллоантигены (например, реакция отторжения трансплантированных органов). Молекулы II класса в норме присутствуют только на В-лимфоцитах и макрофагах крови или тканей.
Молекулы I и II классов лучше рассматривать в качестве сигналов узнавания/программирования для запуска и усиления иммунных реакций в организме. Так, активация цитотоксических Т-лимфоцитов для борьбы с вирусной инфекцией требует присутствия одной и той же молекулы I класса и на инфицированной клетке, и на цитотоксической Т-клетке. Иными словами, «своя» молекула I класса в сочетании с вирусным антигеном формирует новый узнаваемый антиген, на который может реагировать Т-лимфоцит. На клетку, несущую вирусный антиген, но «не свой» антиген HLA I класса, Т-клетка не должна реагировать. Точно так же хелперная Т-клетка активируется только тогда, когда она встречается с антигенпредставляющими клетками (макрофагами), несущими узнаваемую молекулу II класса и антиген, для которого существует точное узнающее место.
Считают, что появление молекул II класса на эндокринных клетках, где они в норме отсутствуют, играет важную роль в аутоиммунном деструктивном процессе, ведущем к возникновению сахарного диабета и других эндокринных заболеваний, таких как тиреоидит Хашимото. Присутствие «своей» молекулы II класса в сочетании с чужеродным или аутоантигеном распознается хелперным Т-лимфоцитом, который затем инициирует активацию иммунной системы, включая образование антител против клетки, несущей сочетание молекулы II класса с чужеродным (или аутологичным) антигеном (см. ниже).
Факторы окружающей среды. Уже отмечалось, что значительное число монозиготных близнецов остается дискордантным по диабету (один близнец с диабетом, а другой — без него). Это свидетельствует о необходимости негенетических факторов для экспрессии диабета у человека. Аналогичные аргументы выдвигает и то обстоятельство, что идентичность гаплотипов HLA не является гарантией конкордантности.
Полагают, что провоцирующим фактором окружающей среды в большинстве случаев служит вирус, способный инфицировать b-клетку. Первоначально вирусную этиологию диабета предполагали, исходя из сезонных колебаний возникновения болезни, а также из-за существования более чем случайной связи между проявлением диабета и предшествующим заболеванием эпидемическим паротитом, гепатитом, инфекционным мононуклеозом, врожденной краснухой и инфицированием вирусом Коксаки. Вирусная гипотеза получила подтверждение в исследованиях, показавших, что некоторые штаммы вируса энцефаломиокардита вызывают диабет у генетически предрасположенных мышей. Выделение вируса Коксаки В4 из поджелудочной железы ранее здорового мальчика, погибшего от приступа кетоацидоза, и индукция диабета у экспериментальных животных, которым инокулировали выделенный вирус, также свидетельствовали о том, что диабет у человека может вызываться вирусами. Повышение титра нейтрализующих антител к вирусу Коксаки за несколько недель до смерти больного указывает на недавнее инфицирование вирусом. В дальнейшем вирусная теория получила свое подтверждение благодаря наблюдениям, согласно которым врожденная краснуха приводит к развитию ИЗСД примерно у 20% пораженных лиц в США. Предположительно, вирусные инфекции могут вызывать диабет двумя путями: в результате непосредственного воспалительного разрушения островков или индукции иммунной реакции.
Однако, несмотря на все сказанное, к вирусной теории надо относиться очень осторожно. Серологические исследования больных с признаками недавней вирусной инфекции и со свежим инсулинзависимым диабетом в лучшем случае дают неопределенные результаты. Если вирусы и служат провоцирующим фактором, то те, которые вызывают острое заболевание, могут и не играть главной роли, а медленно действующие пока не идентифицированы.
Инсулит. У животных активированные Т-лимфоциты инфильтрируют панкреатические островки до или одновременно с развитием диабета. Лимфоциты обнаруживают и в островках поджелудочной железы лиц молодого возраста, погибших от свежего диабета; кроме того, радиоактивно меченные лимфоциты скапливаются в поджелудочной железе больных с ИЗСД. Эти данные согласуются с тем фактом, что иммунные эндокринопатии, как правило, ассоциируются с лимфоцитарной инфильтрацией пораженной ткани. Однако инсулит мог бы быть вторичным феноменом, не имеющим причинной связи с патогенетической последовательностью. На это указывает то обстоятельство, что при экспериментальном диабете у грызунов, вызываемом низкими дозами стрептозотоцина (диабете, имеющем иммунологическую природу), массивную потерю b-клеток регистрирую г до развития инсулита. Больше того, эксперименты на мышах с иммунодефицитом свидетельствуют о том, что участие Т-лимфоцитов необязательно для деструкции b-клеток, вызываемой малыми дозами стрептозотоцина.
Превращениеb-клеткн из «своего» в «не свое» и активация иммунной системы. У больных с инсулинзависимым диабетом повышена частота обнаружения HLA-DR3 и HLA-В15, которые, как известно, ассоциируются с иммунной эндокринопатией. Больше того, ИЗСД часто сочетается с другими формами аутоиммунной эндокринопатии, такими как аддисонова болезнь, тиреоидит Хашимото, гипертиреоз, пернициозная анемия, витилиго, злокачественная миопатия и коллагеноз сосудов (см. гл. 334). Все эти заболевания имеют тенденцию проявляться в определенных семьях. Кроме того, у многих больных инсулинзависимым диабетом в течение первого года после установления диагноза выявляются антитела к клеткам островков Лангерганса. Такие антитела присутствуют и в крови неконкордантных по диабету монозиготных близнецов или тройней, которые заболевают диабетом в будущем. То же справедливо для сиблингов больных инсулинзависимым сахарным диабетом. У 50—60% детей со свежевыявленным диабетом обнаружены Т-клетки-киллеры, причем эта цифра выше, чем в контрольной популяции. Следует отметить, что диабет, сходный с таковым I типа у человека, спонтанно развивается у крыс линии ВВ. У больных животных обнаруживают инсулит, тиреоидит, а также антитела к клеткам панкреатических островков, гладким мышцам, коллоиду щитовидной железы и париетальным клеткам желудка. Диабет у таких животных можно предупредить или вылечить их с помощью иммуномодуляторов.
Что же вызывает аутоиммунный процесс? Во-первых, в крови обнаруживают увеличение отношения Т-хелперов к Т-супрессорам. Это может быть общим феноменом при иммунных эндокринных заболеваниях. Увеличение этого отношения обусловлено, вероятно, дефицитом супрессорных Т-клеток. Несбалансированная популяция хелперных Т-клеток должна была бы служить фактором, предрасполагающим к чрезмерному образованию антител при встрече с антигеном.
Во-вторых, на поверхности b-клеток появляются молекулы HLA II класса. Следует подчеркнуть, что для активации Т-клеток-хелперов требуется присутствие молекул II класса и чужеродного антигена или аутоантигена. Идея заключается в том, что нормальная островковая клетка не экспрессирует молекул II класса, но после внедрения вируса (вероятно, вследствие продукции g-интерферона) такие молекулы на этой клетке появляются, что и делает ее потенциально «не своей». Активация иммунной системы зависит от экспрессируемой аллели. Так, если присутствует HLA-DR2, вероятность возникновения диабета резко снижается, а если присутствуют HLA-DR3 или HLA-DR4 (или, вероятно, DQ-антиген), система должна была бы активироваться. Предполагаемая восприимчивость определяется соответствием между заново появляющимися молекулами II класса, необходимым мембранным антигеном (чужеродным или аутологичным) и конкретной формой Т-клеточного рецептора на хелперной Т-клетке. Это могло бы объяснить развитие ИЗСД при отсутствии HLA-генов высокого риска. Иными словами, в некоторых случаях соответствие между молекулами II класса и Т-клеточным рецептором существует даже в условиях экспрессии обычной аллели низкого риска.
Как это наблюдается и при других опосредованных иммунологическими механизмами эндокринопатиях, признаки активации иммунной системы со временем могут исчезать. Так, антитела к островковым клеткам, присутствующие у впервые выявленных больных с ИЗСД I типа, примерно через год исчезают. Судя по способности секретировать эндогенный инсулин в ответ на пищевой стимул in vivo, присутствие антител к островковым клеткам коррелирует с остаточным количеством b-клеток. По мере исчезновения способности секретировать эндогенный инсулин исчезают и антитела к островковым клеткам. Дело в том, что с гибелью b-клеток исчезает стимул к иммунной реакции.
Разрушение b-клетоки развитие ИЗСД. Поскольку у человека с развивающимся инсулинзависимым диабетом симптомы гипергликемии с полиурией и/или кетоацидозом часто возникают внезапно, долгое время считали, что повреждение b-клеток происходит очень быстро. Однако во многих случаях (в большинстве?) можно наблюдать медленное (в течение нескольких лет) истощение резервов инсулина. Пониманию этого способствовали исследования дискордантности по диабету среди близнецов и тройней, когда у одного из близнецов диабет развивался через много лет после возникновения болезни у другого. При медленном течении болезни самым ранним признаком патологии служит появление антител к островковым клеткам в период, когда уровень сахара в крови еще не превышает нормы, а толерантность к глюкозе остается нормальной. Не меняется и реакция инсулина на нагрузку глюкозой. Затем наступает фаза, когда единственным метаболическим сдвигом является снижение толерантности к глюкозе. Уровень сахара в крови натощак остается нормальным. На третьей стадии развивается гипергликемия натощак, но кетоза не наблюдается даже при плохом контроле диабета. С клинической точки зрения, это инсулиннезависимый сахарный диабет. Однако со временем, особенно в условиях стресса, могут возникать зависимость от инсулина и кетоацидоз. Как отмечалось выше, у многих больных с инсулиннезависимым сахарным диабетом без ожирения на самом деле может быть медленная аутоиммунная форма заболевания.
Иммунологическое разрушение b-клеток происходит, вероятно, как гуморальным, так и клеточно-опосредованным механизмом. Вначале, по-видимому, основную роль играют антитела. Известны два типа антител: цитоплазматические и поверхностные. Обычно у данного больного одновременно присутствуют и те и другие, но могут встречаться и какие-либо одни. Антитела к поверхности островковых клеток способны фиксировать комплемент и лизировать b-клетки. Очевидно, эти антитела нарушают секрецию инсулина еще до того, как b-клетка будет физически повреждена. Они взаимодействуют с мембранным антигеном, который пока точно не охарактеризован. На каком-то этапе начинают действовать цитотоксические Т-лимфоциты и антителозависимые киллерные Т-клетки и деструктивный процесс завершается. К моменту, когда диабет становится явным, большинство инсулинпродуцирующих клеток уже разрушено. По некоторым данным, при диабете I типа масса поджелудочной железы при вскрытии составляла в среднем 40 г (82 г в контроле). Масса эндокринных клеток у лиц с ИЗСД снижалась с 1395 до 413 мг, а масса р-клеток, которая в норме составляет 850 мг, оказалась вообще неопределимой. Поскольку к-клетки остаются в основном интактными, отношение глюкогонпродуцирющих клеток к инсулинпродуцирующим достигало бесконечности.
Дата добавления: 2016-03-05; просмотров: 805;