Таким образом, регуляция АД определяется оптимальным соотношением прессорной и депрессорной систем организма.
1. К прессорной системе относят:
• симпатико‑адреналовую (САС);
• РААС;
• систему антидиуретического гормона (вазопрессин);
• систему простагландина F2a и циклических нуклеотидов;
• эндотелин‑1.
2. Депрессорная система включает:
• аортокаротидную зону (рефлексы с которой ведут к снижению АД);
• систему депрессорных простагландинов;
• калликреин‑кининовую систему (в частности, брадикинин);
• предсердные натрийуретические пептиды;
• эндотелийзависимый релаксирующий фактор (прежде всего оксид азота).
При ГБ имеется рассогласование прессорной и депрессорной систем в виде различных сочетаний повышения активности прессорной и снижения активности депрессорной систем (рис. 2–10).
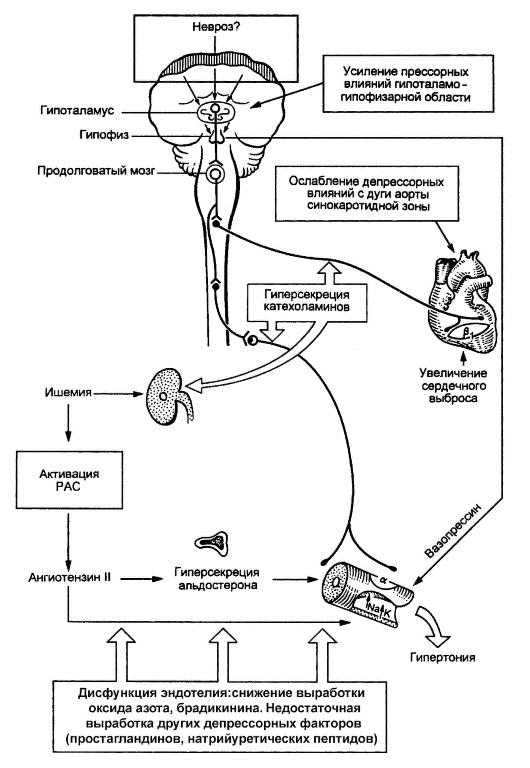
Рис. 2–10. Схематическое изображение патогенеза гипертонической болезни
По не вполне ясным причинам у больных ГБ повышается прессорная активность гипоталамо‑гипофизарной зоны, что ведет к гиперпродукции катехоламинов (повышенная активность симпатико‑адреналовой системы), о чем свидетельствует повышение суточной экскреции с мочой норадреналина, что еще в большей степени возрастает в условиях физического и эмоционального стресса.
Результатом активации симпатико‑адреналовой системы являются следующие изменения, обусловливающие рост АД:
1. периферическая веноконстрикция сопровождается увеличением притока крови к сердцу и сердечного выброса;
2. возрастает число сердечных сокращений, что в сочетании с увеличенным ударным объемом также ведет к увеличению сердечного выброса;
3. возрастает общее периферическое сопротивление сосудов за счет активации р1‑рецепторов периферических артериол (рис. 2–11).
Существенное место среди прессорных факторов занимает активация РААС. Образуемый печенью ангиотензиноген под влиянием ренина, вырабатываемого почкой, трансформируется в ангиотензин I. Ангиотензин I под влиянием АПФ преобразуется в очень мощный прессорный агент – ангиотензин II.
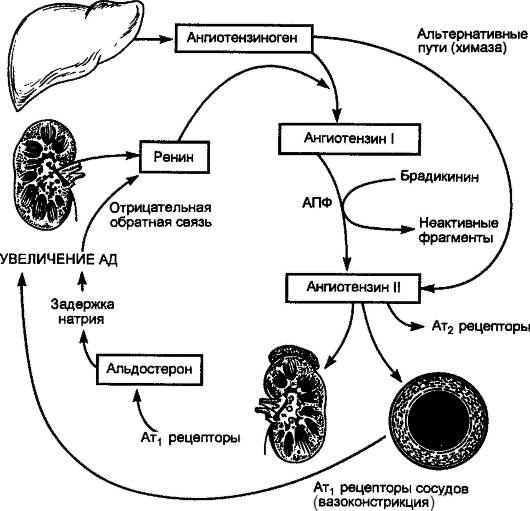
Рис. 2–11. Ренин‑ангиотензиновая система при гипертонической болезни (подробное объяснение в тексте)
Повышенная продукция ренина выступает следствием двух причин:
1. непосредственного воздействия катехоламинов на клетки, вырабатывающие ренин;
2. ишемии почки, обусловленной спазмом почечных сосудов под влиянием катехоламинов, что ведет к гипертрофии и гиперплазии юкстагломерулярного аппарата (ЮГА), вырабатывающего ренин.
Повышенное содержание ангиотензина II в плазме крови вызывает длительный спазм гладкой мускулатуры периферических артериол и резкое увеличение общего периферического сосудистого сопротивления.
Роль ангиотензина II в патогенезе ГБ исключительно велика, так как кроме прямого прессорного влияния он обусловливает развитие и других патологических процессов – гипертрофию и фиброз миокарда левого желудочка, гипертрофию гладких мышечных волокон сосудов, способствует развитию нефросклероза, повышению реабсорбции натрия и воды, высвобождению катехоламинов из мозгового слоя надпочечников. Весьма существенно, что кроме повышения уровня ангиотензина II в кровяном русле повышается его содержание в тканях, так как существуют так называемые тканевые ренин‑ангиотензиновые системы. Наконец, кроме классического пути образования ангиотензина II путем воздействия АПФ на ангиотензин I, существуют так называемые альтернативные пути, когда ангиотензин I превращается в ангиотензин II с помощью других ферментов (например, химазы), а также нерениновый путь образования ангиотензина II.
Ангиотензин II оказывает влияние и на другие прессорные системы:
1. вызывая жажду, он ведет к повышенной выработке вазопрессина, обусловливающего спазм сосудов и задержку жидкости в организме;
2. активирует выработку альдостерона – гормона коры надпочечников, обусловливающего задержку в организме натрия и воды (увеличение массы циркулирующей крови);
3. ангиотензин II также обладает пролиферативным влиянием на клетки гладкой мускулатуры сосудов, обусловливая изменение их структуры (так называемое ремоделирование сосудов), что в еще большей мере ведет к росту общего периферического сосудистого сопротивления.
Длительному спазму артериол способствует повышенное содержание ионов Са2+ в цитозоле гладкомышечных волокон, что связано с наследственно обусловленными особенностями транспорта ионов через полупроницаемые мембраны.
Повышение активности прессорных факторов сочетается с ослаблением депрессорных влияний с дуги аорты и синокаротидной зоны, уменьшением выработки кининов, недостаточной активацией выработки предсердного натрийуретического и эндотелийзависимого релаксирующего факторов (оксида азота), уменьшением выделения простагландинов, обладающих депрессорным влиянием, и простациклина, уменьшением выработки ингибитора ренина – фосфолипидного пептида. Снижение выработки депрессорных факторов связывают с так называемой эндотелиальной дисфункцией, когда под влиянием ряда факторов (в частности, АГ) эндотелий начинает продуцировать преимущественно прессорные факторы.
Большое значение в развитии АГ имеют снижение чувствительности тканей к действию инсулина и связанная с этим гиперинсулинемия.
Следует помнить, что АД становится стабильно повышенным тогда, когда развивается так называемое ремоделирование периферических (резистивных) сосудов – уменьшение просвета сосуда в результате увеличения комплекса «интима‑медиа», что может являться следствием пролиферативного эффекта ангиотензина II.
Независимо от преобладающего нейрогуморального механизма повышения АД, развивается поражение «органов‑мишеней» – сердца (гипертрофия и фиброз миокарда с изменением формы левого желудочка – так называемое ремоделирование сердца), сосудов (гипертрофия гладких мышечных волокон с последующим изменением соотношения медиа‑просвет сосуда), артериолосклерозом почек (нефроангиосклероз). Именно от состояния этих органов зависят течение и исход ГБ.
Дата добавления: 2016-02-02; просмотров: 3643;