Электрохимические методы анализа
Физико-химические методы анализа (ФХМА) основаны на использовании зависимости между измеряемыми физическими свойствами веществ и их качественным и количественным составом. Поскольку физические свойства веществ измеряются с помощью различных приборов – «инструментов», то эти методы анализа называют также инструментальными методами.
Наибольшее практическое применение среди ФХМА имеют:
- электрохимические методы – основаны на измерении потенциала, силы тока, количества электричества и других электрических параметров;
- спектральные и другие оптические методы – основаны на явлениях поглощения или испускания электромагнитного излучения (ЭМИ) атомами или молекулами вещества;
- хроматографические методы – основаны на сорбционных процессах, протекающих в динамических условиях при направленном перемещении подвижной фазы относительно неподвижной.
К достоинствам ФХМА можно отнести высокую чувствительность и низкий предел обнаружения – массовый до 10-9 мкг и концентрационный до 10-12 г/мл, высокую селективность (избирательность), позволяющую определять компоненты смесей без их предварительного разделения, а также экспрессность проведения анализов, возможность их автоматизации и компьютеризации.
В аналитической химии широко применяются электрохимические методы. Выбор метода анализа конкретного объекта анализа определяется многими факторами, в том числе, в первую очередь, нижним пределом определения элемента.
Данные о нижнем пределе обнаружения различных элементов некоторыми методами представлены в таблице.
Таблица
Пределы определения (мкг/мл) элементов различными методами
| Элемент | МАС | ААС | ПТП | ИВА | Ионо- метрия | Ампером.титров. |
| Ag | 0,1– дитизон | 0,07 | 0,2 | 0.00001 | 0.02 | 0.05 |
| As | 0,05 - молибд.синь | 0,2 | 0,04 | 0,02 | - | 0,05 |
| Au | 0,04-метил.фиол. | 0,3 | 0,005 | 0,001 | - | 0,05 |
| Bi | 0,07-дитизон | 0,005 | 0,00001 | - | 0,5 | |
| Cd | 0,04-дитизон | 0,05 | 0,002 | 0,00001 | 0,03 | 0,5 |
| Cr | 0,04-дифе-нилкарбазид | 0,2 | 0,02 | - | - | |
| Cu | 0,03-дитизон | 0,2 | 0,002 | 0,00002 | 0,01 | 0,05 |
| Hg | 0,08-дитизон | - | 0,00005 | |||
| Pb | 0,08-дитизон | 0,6 | 0,003 | 0,00002 | 0,03 | |
| Sb | 0,08-родамин | 0,004 | 0,00004 | - | 0,5 | |
| Fe | 0,1-роданид | 0,2 | 0,003 | 0,0002 | 0,3 | 0,5 |
| Se | 0,08-диами-нофталин | 0,3 | 0,2 | 0,00002 | - | 0,5 |
| Sn | 0,07-фенил-флуриом | 0,4 | 0,003 | 0,00004 | - | 0,5 |
| Te | 0,1-висмутол | 0,7 | 0,02 | - | - | |
| Tl | 0,06-родамин | 0,6 | 0,01 | 0,00002 | - | 0,5 |
| Zn | 0,02-дитизон | 0,02 | 0,003 | 0,0003 | - | 0,5 |
| F- | - | - | - | - | 0,02 | 5-10 |
| NH4+,NO3- | - | - | - | - | 0,1 | 1-5 |
МАС - молекулярная абсорбционная спекрометрия (фотометрия);
ААС - атомно-абсорбционная спектрометрия (пламенная фотометрия);
ПТП - переменно-токовая полярография;
ИВА - инверсионная вольтамперометрия.
Погрешности определений в ФХМА составляют около 2-5%, проведение анализов требует применения сложной и дорогостоящей аппаратуры.
Различают прямые и косвенные методы физико-химического анализа. В прямых методах используют зависимость величины измеряемого аналитического сигнала от концентрации определяемого компонента. В косвенных методах аналитический сигнал измеряют с целью нахождения конечной точки титрования определяемого компонента подходящим титрантом, то есть используют зависимость измеряемого параметра от объѐматитранта.
Электрохимические методы анализа основаны на изучении и использовании процессов, протекающих на поверхности электрода или в приэлектродном пространстве. Любой электрический параметр (потенциал, электрический ток, количество электричества и др.), функционально связанный с концентрацией определяемого компонента и поддающийся правильному измерению, может служить аналитическим сигналом.
По природе измеряемого аналитического сигнала электрохимические методы анализа разделяют на потенциометрию, вольтамперометрию, кулонометрию и ряд других методов:
Характеристическая зависимость электрохимического сигнала от независимой переменной
| Метод | Измеряемый сигнал | Зависимость сигнала от независимой переменной |
| Потенциометрия, ионометрия | потенциал E = f(C) С-концентрация анализируемого вещества | 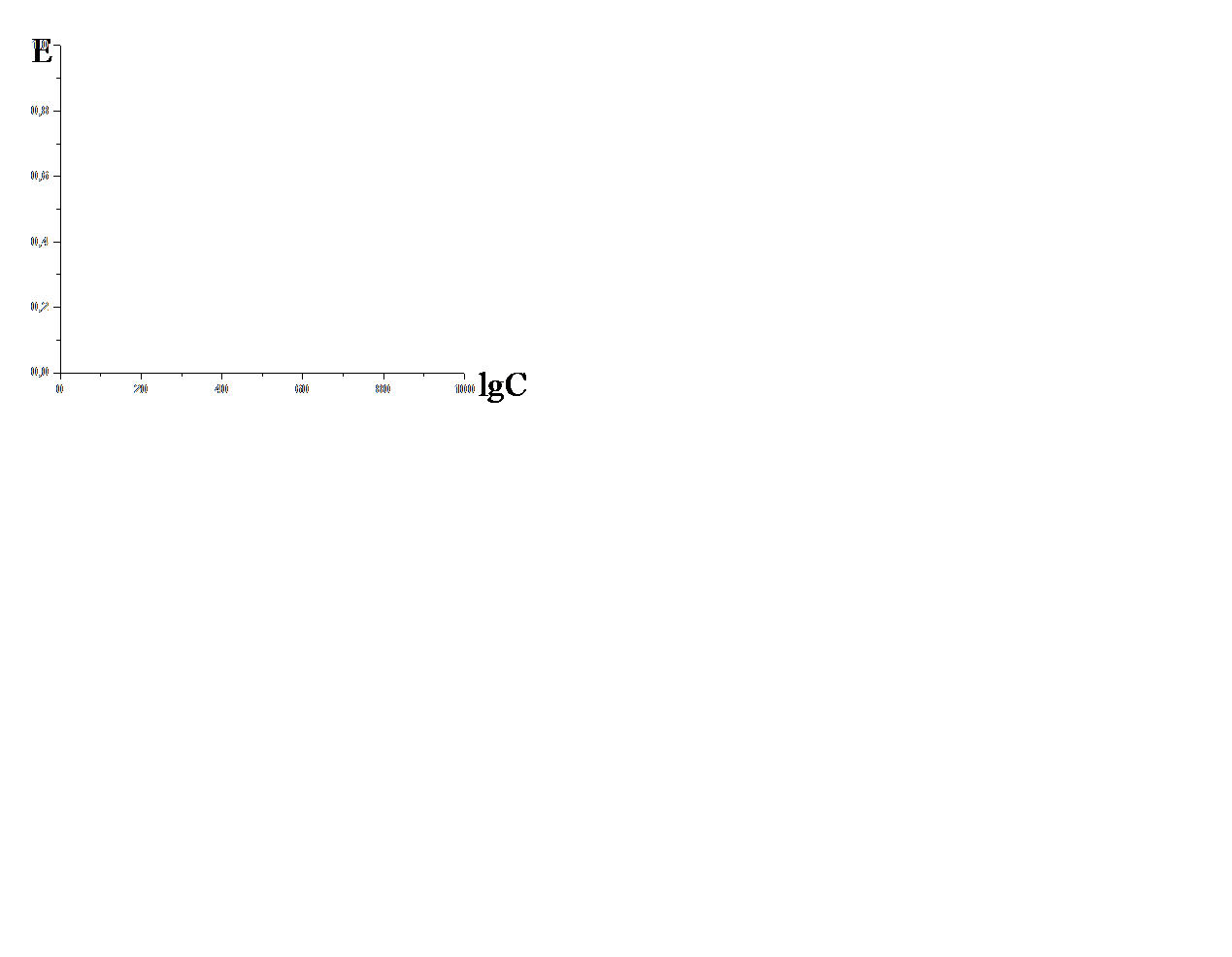
|
| Потенциометрическое титрование | потенциал E = f(V), V- объем реагента-титранта |
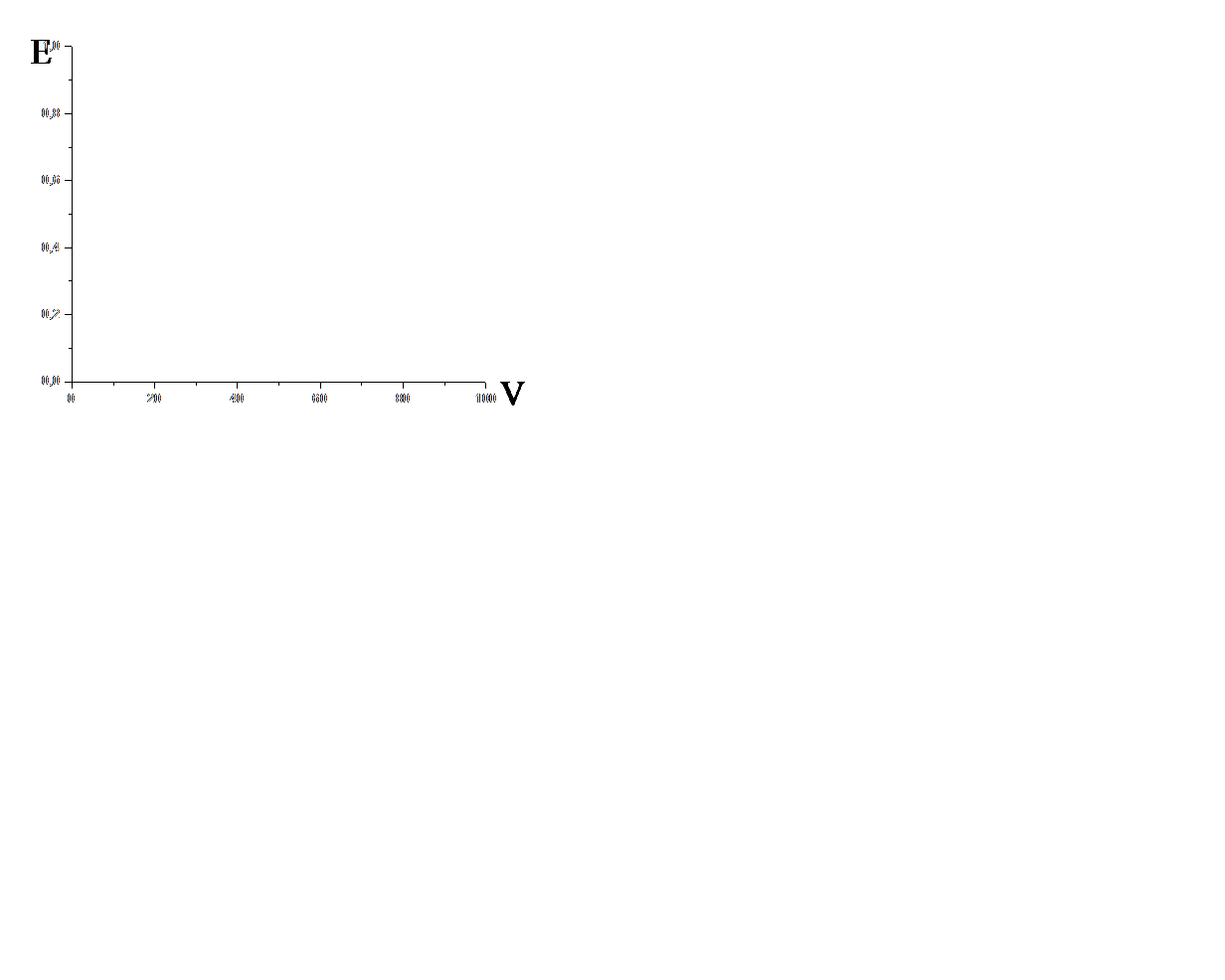
|
| полярография, вольтамперометрия | ток I = f(E), E – потенциал поляризации электрода | 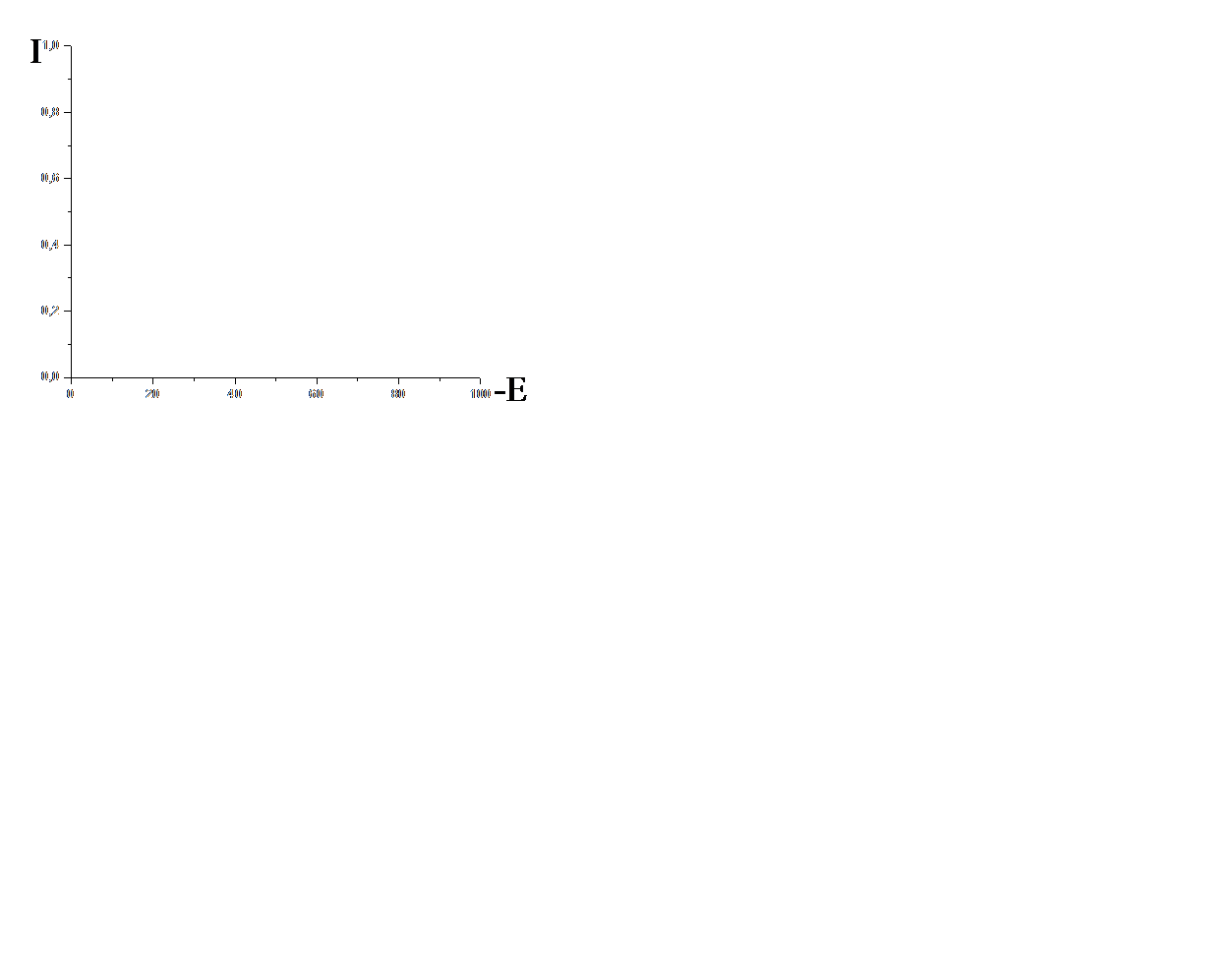
|
| инверсионная вольтамперометрия | ток In = f(E) | 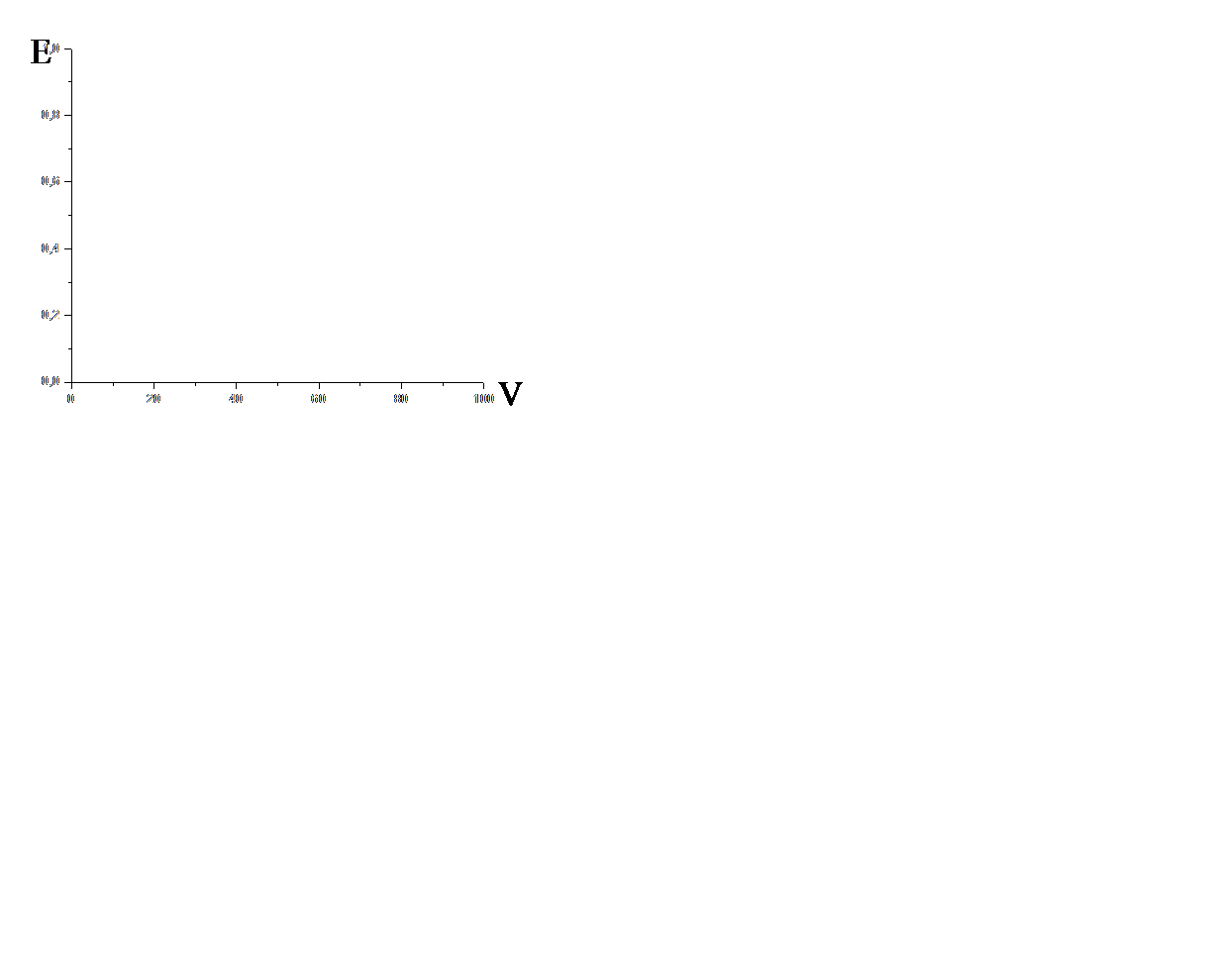
|
| хронопотенциометрия | потенциал E =f(t), t – время поляризации электрода при I=const. |
|
| амперометрическое титрование с одним индикаторным электродом | ток I = f(V), V – объем реагента-титранта | 
|
| амперометрическое титрование с двумя индикаторными электродами | ток I = f(V) V – объем реагента-титранта |
|
| кулонометрия | Q = f(C), С – количество вещества | 
|
| кондуктометрия | G = f(C), С – концентрация ионов в растворе | 
|
| кондуктометрическое титрование | электропроводность G = f(V), V – объем реагента-титранта | 
|
Потенциометрия
В основе потенциометрических измерений лежит зависимость равновесного потенциала электрода от активности (концентрации) определяемого иона. Для измерений необходимо составить гальванический элемент из подходящего индикаторного электрода и электрода сравнения, а также иметь прибор для измерения потенциала индикаторного электрода (ЭДС гальванического элемента), в условиях близких к термодинамическим, когда индикаторный электрод имеет равновесный (или близкий к нему) потенциал, то есть без отвода заметного тока от гальванического элемента при замыкании цепи. При этом нельзя использовать обычный вольтметр, а следует применять потенциометр - электронный прибор с большим входным сопротивлением (1011 - 1012 Ом), что исключает протекание электродных электрохимических реакций и возникновение тока в цепи.
Индикаторный электрод – это электрод, потенциал которого зависит от активности (концентрации) определяемого иона в анализируемом растворе.
Электрод сравнения – это электрод, потенциал которого в условиях проведения анализа остается постоянным. По отношению к электроду сравнения измеряют потенциал индикаторного электродаЕ(ЭДС гальванического элемента).
В потенциометрии используют два основных класса индикаторных электродов – электронообменные и ионообменные.
Электронообменныеэлетроды – это электроды, на поверхности которых протекают электродные реакции с участием электронов. К таким электродам относятся электроды первого и второго рода, окислительно-восстановительные электроды.
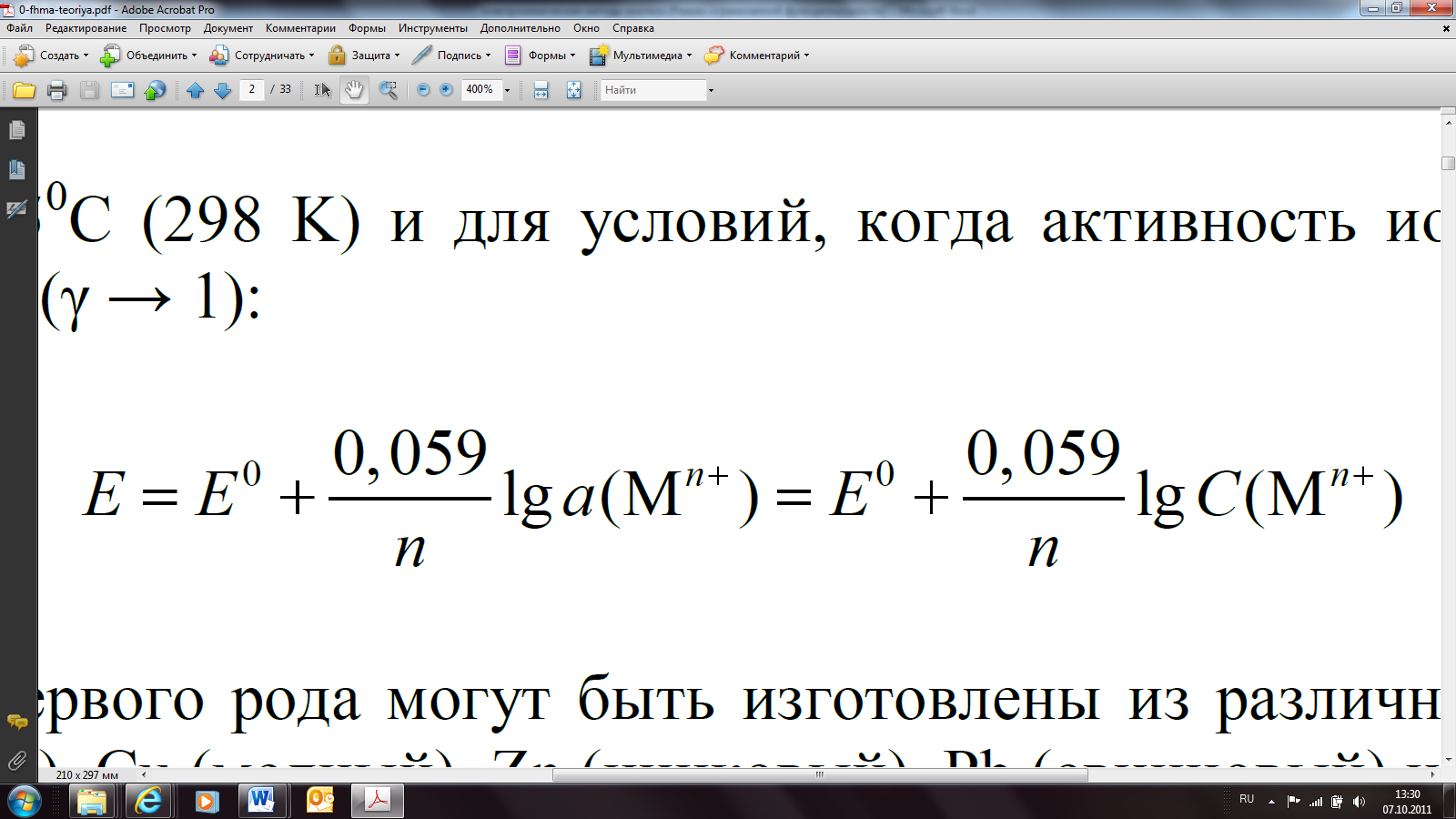
Электроды первого рода – это электроды, обратимые по катиону, общему с материалом электрода, например, металл М, погруженный в раствор соли того же металла. На поверхности такого электрода протекает обратимая реакция Mn+ + ne ↔ M и его реальный потенциал зависит от активности (концентрации) катионов металла в растворе в соответствии с уравнением Нернста:

Для температуры 250С (298 K) и для условий, когда активность ионов приблизительно равна концентрации (γ → 1):
Электроды первого рода могут быть изготовлены из различных металлов, например, Ag (серебряный), Cu (медный), Zn (цинковый), Pb (свинцовый) и др.
Схематически электроды первого рода записывают как М | Mn+, где вертикальной линией показана граница твердой (электрод) и жидкой (раствор) фаз. Например, серебряный электрод, погруженный в раствор нитрата серебра изображают следующим образом – Ag | Ag+; при необходимости указывают концентрацию электролита – Ag | AgNO3 (0,1 M).
К электродам первого рода относится и газовый водородный электродPt(H2) | H+ (2Н+ + 2е ↔ Н2, Е0 = 0):
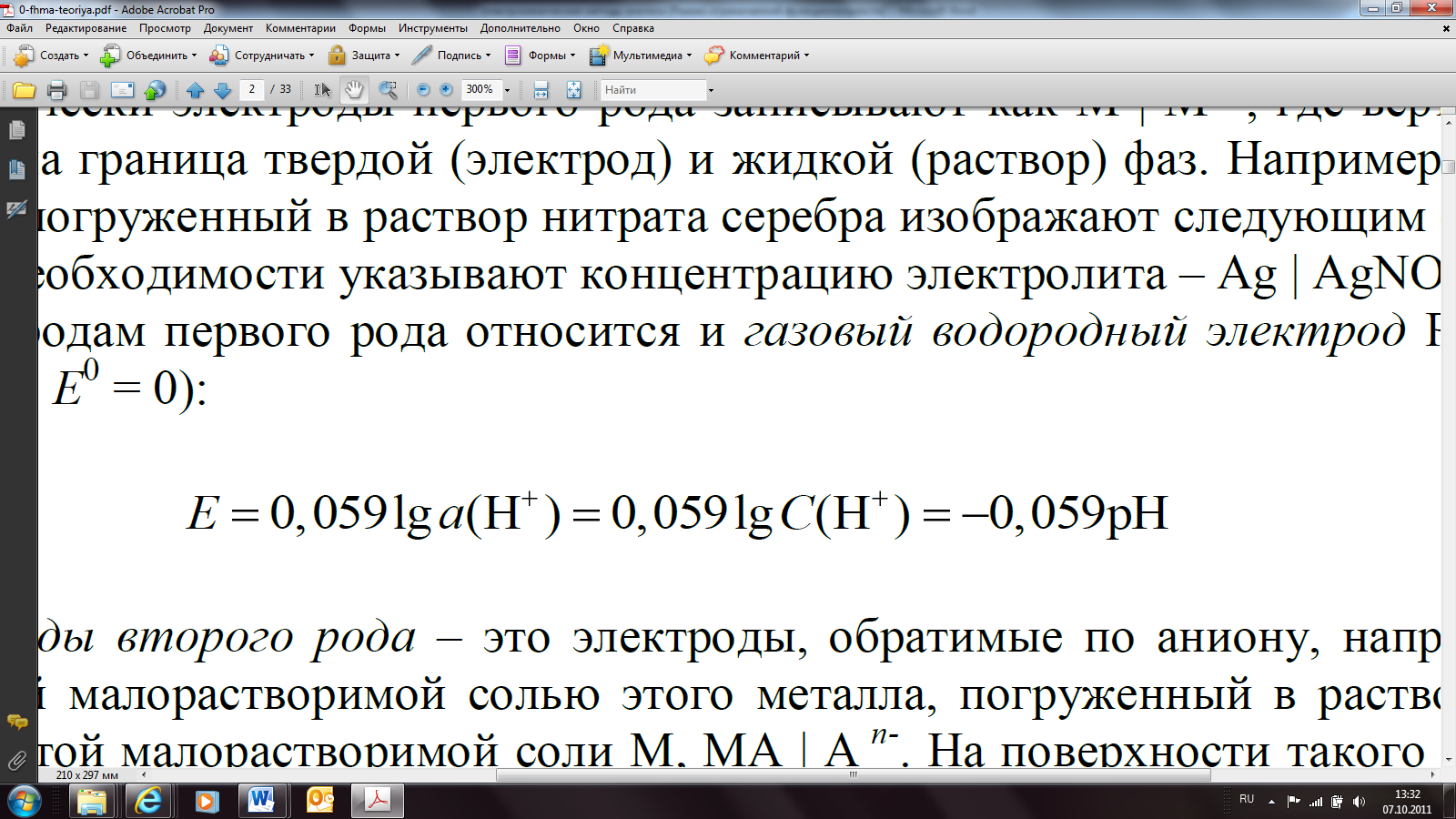
Электроды второго рода – это электроды, обратимые по аниону, например, металл, покрытый малорастворимой солью этого металла, погруженный в раствор, содержащий анион этой малорастворимой соли M, MA | Аn-. На поверхности такого электрода протекает обратимая реакция MА + ne ↔ M + Аn- и его реальный потенциал зависит от активности (концентрации) аниона в растворе в соответствии с уравнением Нернста (приТ= 298 K и γ → 1):

Примерами электродов второго рода служат хлорсеребряный (AgCl + e ↔ Ag + Cl-) и каломельный (Hg2Cl2 + 2e ↔ 2Hg + 2Cl-) электроды:

Окислительно-восстановительные электроды – это электроды, которые состоят из инертного материала (платина, золото, графит, стеклоуглерод и др.), погруженного в раствор, содержащий окисленную (Ок) и восстановленную (Вос) формы определяемого вещества. На поверхности такого электрода протекает обратимая реакция Ок + ne ↔ Вос и его реальный потенциал зависит от активности (концентрации) окисленной и восстановленной форм вещества в растворе в соответствии с уравнением Нернста (приТ= 298 K и γ → 1):

Если в электродной реакции участвуют ионы водорода, то их активность (концентрацию) учитывают в соответствующих уравнениях Нернста для каждого конкретного случая.
Ионообменные электроды – это электроды, на поверхности которых протекают ионообменные реакции. Такие электроды называют также ионселективными или мембранными.Важнейшей составной частью таких электродов является полупроницаемая мембрана – тонкая твердая или жидкая пленка, отделяющая внутреннюю часть электрода (внутренний раствор) от анализируемого и обладающая способностью пропускать только ионы одного вида Х (катионы или анионы). Конструктивно мембранный электрод состоит из внутреннего электрода сравнения (обычно хлорсеребряный) и внутреннего раствора электролита с постоянной концентрацией потенциалопределяющего иона, отделенных от внешнего (исследуемого) раствора чувствительной мембраной.
Реальный потенциал ионселективных электродов, измеренный относительно какого-либо электрода сравнения, зависит от активности тех ионов в растворе, которые сорбируются мембраной:

где const – константа, зависящая от природы мембраны (потенциал асимметрии) и разности потенциалов внешнего и внутреннего электродов сравнения, n иа(Хn±) – заряд и активность потенциалопределяющего иона. Если потенциал ионселективного электрода измерен относительно стандартного водородного электрода, то константа является стандартным электродным потенциалом Е0.
Для мембранных электродов значение крутизны электродной функции может отличаться от теоретической нернстовской величины (0,059 В); в этом случае реальное значение электродной функции θ определяют как тангенс угла наклона градуировочного графика. Тогда:

Потенциал мембранного электрода в растворе, содержащем кроме определяемого иона Х посторонний ион В, влияющий на потенциал электрода, описывается уравнением Никольского (модифицированное уравнение Нернста):

где z – заряд постороннего (мешающего) иона, KХ/В– коэффициент селективности мембранного электрода.
Коэффициент селективности KХ/В характеризует чувствительность мембраны электрода к определяемым ионам Х в присутствии мешающих ионов В. Если KХ/В<1, то электрод селективен относительно ионов Х и, чем меньше числовое значение коэффициента селективности, тем выше селективность электрода по отношению к определяемым ионам и меньше мешающее действие посторонних ионов. Если коэффициент селективности равен 0,01, то это означает, что мешающий ион В оказывает на величину электродного потенциала в 100 раз меньшее влияние, чем определяемый ион той же молярной концентрации.
Рассчитывают коэффициент селективности как отношение активностей (концентраций) определяемого и мешающего ионов, при которых электрод приобретает одинаковый потенциал в растворах этих веществ, с учѐтом их зарядов:

Зная значение коэффициента селективности можно рассчитать концентрацию мешающего иона, влияющую на потенциал ионселективного электрода (пример).
Пример. Какую концентрацию нитратных ионов нужно создать в 1∙10-3 М растворе фторида натрия, чтобы ионселективный фторидный электрод был одинаково чувствителен к обоим ионам, если его коэффициент селективности электрода?

Решение.
Так как , то
Это означает, что концентрация нитратных ионов в анализируемом растворе свыше 0,5 моль/л оказывает значительное влияние на определение фторид-иона в его миллимо-лярных растворах.
Классическим примером ионселективного электрода с твердой мембраной является стеклянный электрод с водородной функцией, служащий для измерения концентрации ионов водорода в растворе (стеклянный рН-электрод). Для таких электродов мембраной служит специальное стекло определѐнного состава, а внутренним электролитом – 0,1 М раствор хлороводородной кислоты:
Ag, AgCl | 0,1 M HCl | стеклянная мембрана | исследуемый раствор
На поверхности стеклянной мембраны происходит ионообменный процесс:
-SiO-Na+ (стекло) + Н+ (раствор) → -SiO-H+ (стекло) + Na+ (раствор)
в результате чего устанавливается динамическое равновесие между ионами водорода в стекле и растворе Н+ (стекло) ↔ Н+ (раствор), что приводит к возникновению потенциала:
E = const + θlga(H+) = const – θpH
Стеклянный электрод с повышенным содержанием в мембране Al2O3 измеряет ак-тивность ионов натрия в растворе (стеклянный Na-электрод, натрийселективныйэлек-трод). В этом случае внутренним раствором служит 0,1 М раствор хлорида натрия:
Ag, AgCl | 0,1 M NaCl | стеклянная мембрана | исследуемый раствор
На поверхности стеклянной мембраны натрийселективного электрода устанавливается равновесие между ионами натрия в стекле и растворе Na+ (стекло) ↔ Na+ (раствор), что приводит к возникновению потенциала:
E = const + θlga(Na+) = const – θpNa
Наиболее совершенным электродом с кристаллической мембраной является фторидселективный электрод, мембрана которого выполнена из пластинки монокристалла фторида лантана (LaF3), активированного для увеличения проводимости фторидом европия (EuF2):
Ag, AgCl | 0,1 M NaCl, 0,1 M NaF | LaF3 (EuF2) | исследуемый раствор
Потенциал фторидного электрода определяется ионообменным процессом на его поверхности F- (мембрана) ↔ F- (раствор):
E = const – θlga(F-) = const + θpF
Значения константы и крутизны электродной функции θ для ионселективных электродов определяют из градуировочного графикаЕ ÷ рХ как отрезок на оси ординат и тангенс угла наклона прямой соответственно. Для стеклянного рН-электрода эта операция заменяется настройкой приборов (рН-метров) по стандартным буферным растворам с точно известными значениями рН.
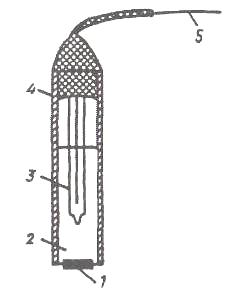
Схематический вид стеклянного и фторидселективного электродов приведены на рисунках:
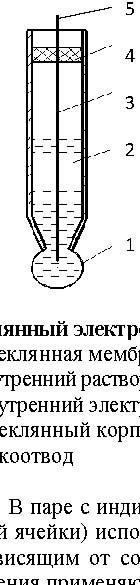
| 
|
| Стеклянный электрод | Фторидселективный электрод |
| 1 – стеклянная мембрана | 1 - пластинка из LaF3 (EuF2) |
| 2 - внутренний раствор (HCl или NaCl) | 2 - внутренний раствор (NaCl+NaF) |
| 3– внутренний электрод сравнения | 3 - внутренний электрод сравнения |
| 4 - стеклянный корпус | 4 – пластиковый корпус |
| 5 – токоотвод | 5 – токоотвод |
В паре с индикаторным электродом для измерения его потенциала (ЭДС гальванической ячейки) используют электрод сравнения с известным и устойчивым потенциалом, не зависящим от состава исследуемого раствора. Наиболее часто в качестве электрода сравнения применяют хлорсеребряный и каломельный электроды. Оба электрода относятся к электродам второго рода и характеризуются высокой стабильностью в работе.
Потенциалы хлорсеребряного и каломельного электродов зависят от активности (концентрации) хлорид-ионов (приТ= 298 K и γ → 1):

В качестве электродов сравнения чаще всего применяют электроды с насыщенным раствором хлорида калия – при 250С потенциал насыщенного хлорсеребряного электрода сравнения равен +0,201 В, а насыщенного каломельного +0,247 В (относительно стандартного водородного электрода). Потенциалы для хлорсеребряных и каломельных электродов сравнения, содержащих 1 М и 0,1 М растворы хлорида калия, можно найти в справочных таблицах.
Схематический вид насыщенных хлорсеребряного и каломельного электродов срав-нения приведены на рисунке:
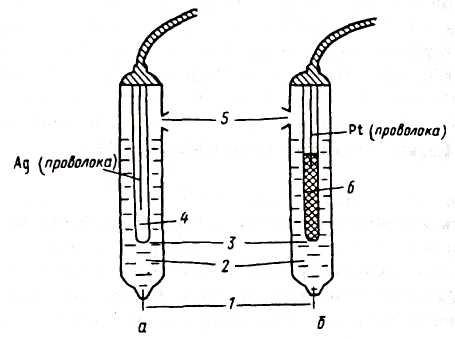
Электроды сравнения хлорсеребряный (а) и каломельный (б)
1 - асбестовое волокно, обеспечивающее контакт с анализируемым раствором
2 - раствор KCl (насыщенный)
3 - отверстие для контакта
4 - раствор KCl (насыщенный), AgCl (тв.)
5 - отверстие для ввода раствора KCl
6 - паста из смеси Hg2Cl2, Hg и КС1 (насыщенный)
Потенциометрический анализ широко применяют для непосредственного определения активности (концентрации) ионов в растворе путем измерения равновесного потенциала индикаторного электрода (ЭДС гальванического элемента) – прямая потенциометрия (ионометрия), а также для индикации конечной точки титрования (ктт) по изменению потенциала индикаторного электрода в процессе титрования (потенциометрическое титрование).
Во всех приемахпрямой потенциометрии используется зависимость индикаторного электрода от активности (концентрации) определяемого иона, которая описывается уравнением Нернста. Результаты анализа подразумевают определение концентрации вещества, а не его активности, что возможно при значении коэффициентов активности ионов равных единице (γ → 1) или их постоянном значении (постоянной ионной силе раствора), поэтому в дальнейших рассуждениях используются только концентрационные зависимости.
Концентрация определяемого иона может быть рассчитана по экспериментально найденному потенциалу индикаторного электрода, если для электрода известны постоянная составляющая (стандартный потенциал Е0) и крутизна электродной функции θ. В этом случае составляется гальванический элемент, состоящий из индикаторного электрода и электрода сравнения, измеряется его ЭДС, рассчитывается потенциал индикаторного электрода (относительно СВЭ) и концентрация определяемого иона.
В методеградуировочного графика готовят серию стандартных растворов с известной концентрацией определяемого иона и постоянной ионной силой, измеряют потенциал индикаторного электрода относительно электрода сравнения (ЭДС гальванического элемента) в этих растворах и по полученным данным строят зависимость Е ÷ рС(А) (градуировочный график). Затем измеряют потенциал индикаторного электрода в анализируемом растворе Ех (в тех же условиях) и по графику определяют рСх(А) и рассчитывают концентрацию определяемого вещества в анализируемом растворе.
В методе стандарта (сравнения) измеряют потенциал индикаторного электрода в анализируемом растворе (Ех) и в стандартном растворе определяемого вещества (Ест). Расчет концентрации определяемого иона проводят исходя из уравнений Нернста для анализируемой пробы и стандартного образца. Крутизна электродной функции для индикаторного электрода θ должна быть известна или определена заранее по градуировочному графику.
При использовании метода добавок сначала измеряют потенциал индикаторного электрода в анализируемом растворе (Ех), затем добавляют к нему определенный объём стандартного раствора определяемого вещества и измеряют потенциал электрода в полученном растворе с добавкой (Ех+д). Расчет концентрации определяемого иона проводят исходя из уравнений Нернста для анализируемой пробы и пробы с добавкой. Крутизна электродной функции для индикаторного электрода θдолжна быть известна или определена заранее по градуировочному графику.
При потенциометрическом титровании измеряют и записывают ЭДС электрохимической ячейки (потенциал индикаторного электрода) после добавления каждой порции титранта. Затем по полученным результатам строят кривые титрования – интегральную в координатах E ÷ V(а) и дифференциальную в координатах ∆E/∆V ÷ V (б), и определяют конечную точку титрования (ктт) графическим способом:
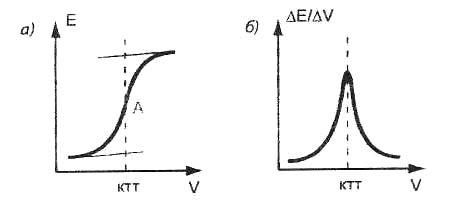
В потенциометрическом титровании используют все основные типы химических реакций – кислотно-основные, окислительно-восстановительные, осаждения и комплексообразования. К ним предъявляются те же требования, что и в визуальной титриметрии, дополненные наличием подходящего индикаторного электрода для фиксации изменения концентрации потенциалопределяющих ионов в ходе титрования.
Погрешность определения при проведении потенциометрического титрования составляет 0,5-1%, что существенно ниже, чем при прямых потенциометрических измерениях (2-10%), однако, при этом наблюдаются более высокие пределы обнаружения – больше 10-4 моль/л.
Кулонометрия
Кулонометрия объединяет методы анализа, основанные на измерении количества электричества, затраченного на электрохимическую реакцию. Электрохимическая реакция приводит к количественному электропревращению (окислению или восстановлению) определяемого вещества на рабочем электроде (прямая кулонометрия) или к получению промежуточного реагента (титранта), который стехиометрически реагирует с определяемым веществом (косвенная кулонометрия, кулонометрическое титрование).
В основе кулонометрических методов лежит закон Фарадея, который устанавливает связь между количеством электропревращенного (окисленного или восстановленного) вещества и количеством израсходованного при этом электричества:

где m – масса электропревращенного вещества,г; Q – количество электричества, затраченного на электропревращение вещества, Кл; F – число Фарадея, равное количеству электричества, необходимого для электропревращения одного моль-эквивалента вещества, 96500 Кл/моль; М – молярная масса вещества, г/моль; n – число электронов, участвующих в электрохимической реакции.
Необходимым условием проведения кулонометрического анализа является практически полное расходование электричества на превращение определяемого вещества, то есть электрохимическая реакция должна протекать без побочных процессов со 100% вы-ходом по току.
На практике кулонометрический анализ реализуется в двух вариантах – при постоянном потенциале (потенциостатическаякулонометрия) и при постоянной силе тока(амперостатическаякулонометрия).
Потенциостатическуюкулонометрию применяют для прямых кулонометрических измерений, когда электролизу подвергается непосредственно определяемое вещество. При этом потенциал рабочего электрода с помощью потенциостатов поддерживается постоянным и его значение выбирают на основе поляризационных кривых в области предельного тока определяемого вещества. В процессе электролиза при постоянном потенциале сила тока уменьшается в соответствии с уменьшением концентрации электроактивного вещества по экспоненциальному закону:
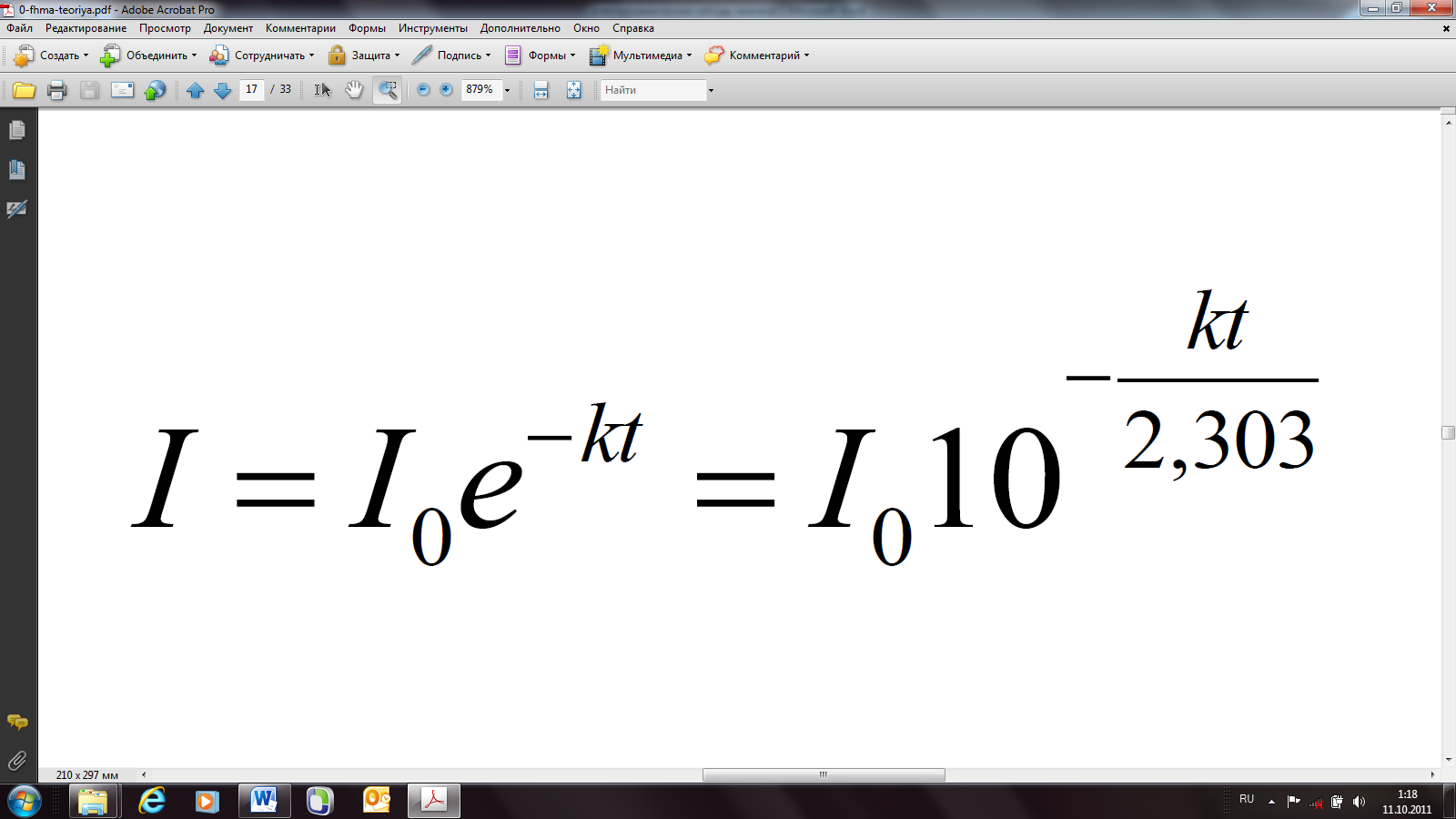
где Ι – сила тока в момент времени t, А; Ι0 – сила тока в начальный момент электролиза, А; k – константа, зависящая от условий электролиза.
Электролиз ведут до достижения остаточного тока Ι, величина которого определяется требуемой точностью – для допустимой погрешности 0,1% электролиз можно считать завершенным при Ι = 0,001Ι0. Для сокращения времени электролиза следует применять рабочий электрод большой поверхности при интенсивном перемешивании анализируемого раствора.
Общее количество электричества Q, необходимое для электропревращения определяемого вещества, определяется уравнением:
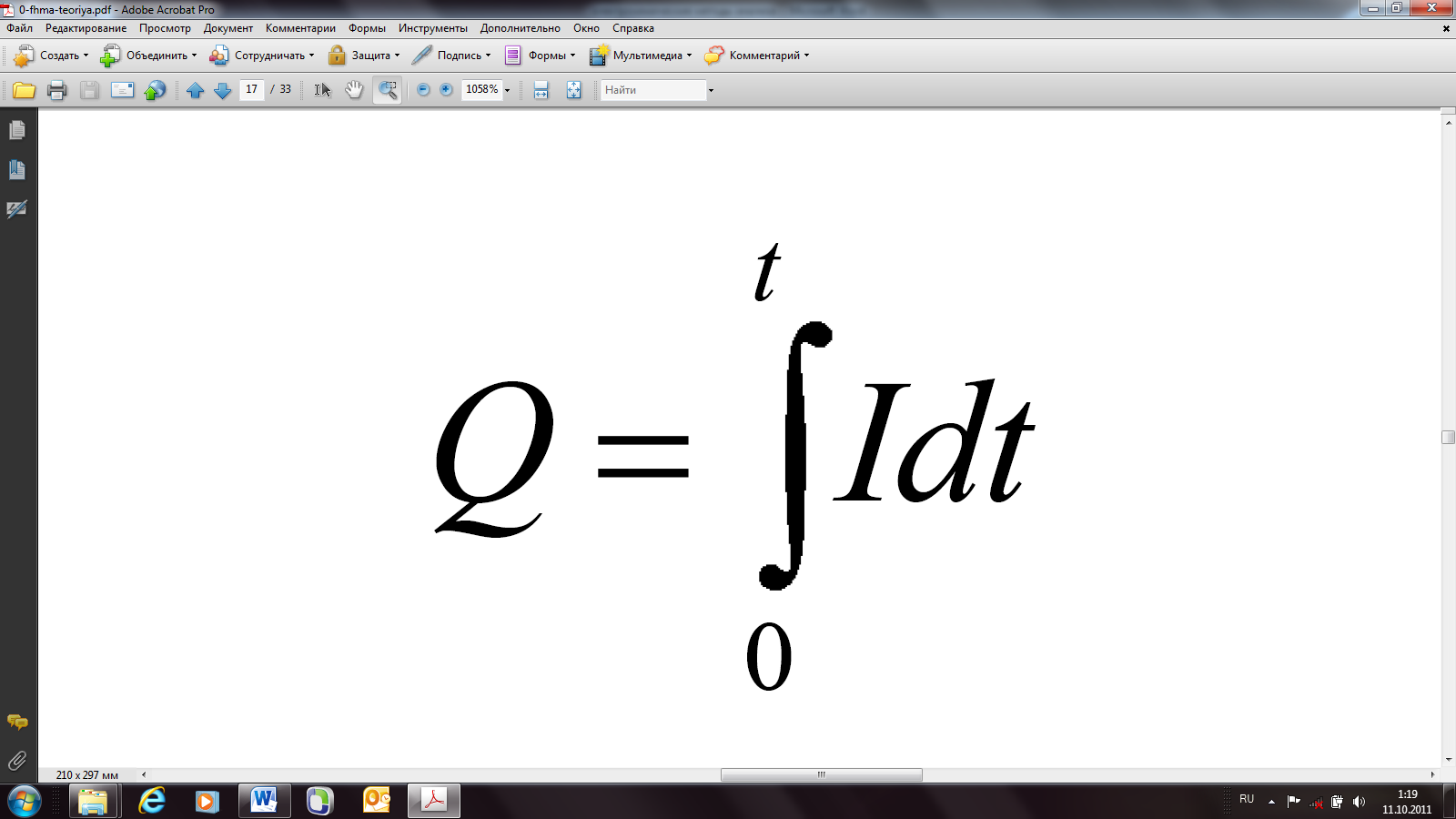
Определить количество электричества можно измерением площади под кривой «ток – время» с помощью механических или электронных интеграторов, либо с помощью химических кулонометров. Кулонометр – это электролитическая ячейка, в которой со 100% выходом по току протекает электрохимическая реакция известной стехиометрии. Кулонометр включают последовательно с исследуемой кулонометрической ячейкой, поэтому за время электролиза через обе ячейки протекает одинаковое количество электричества. Если по окончании электролиза измерить количество (массу) образовавшегося в кулонометре вещества, то по закону Фарадея можно рассчитать количество электричества. Чаще всего применяют серебряный, медный и газовые кулонометры.
Амперостатическую кулонометрию применяют для кулонометрического титрования при постоянном токе, в процессе которого определяемое вещество реагирует с титрантом, образующимся в результате электрохимической реакции на рабочем электроде, а потому, называемый электрогенерированным титрантом.
Для обеспечения 100%-ного выхода по току необходим значительный избыток вспомогательного вещества, из которого генерируется титрант, что исключает протекание конкурирующих реакций на рабочем электроде. При этом титрант генерируется в количестве, эквивалентном определяемому веществу, и по количеству электричества, затраченного на генерацию титранта, можно рассчитать содержание определяемого вещества.
Количество электричества Q в кулонометрии при постоянной силе тока Ι рассчитывают по формуле:
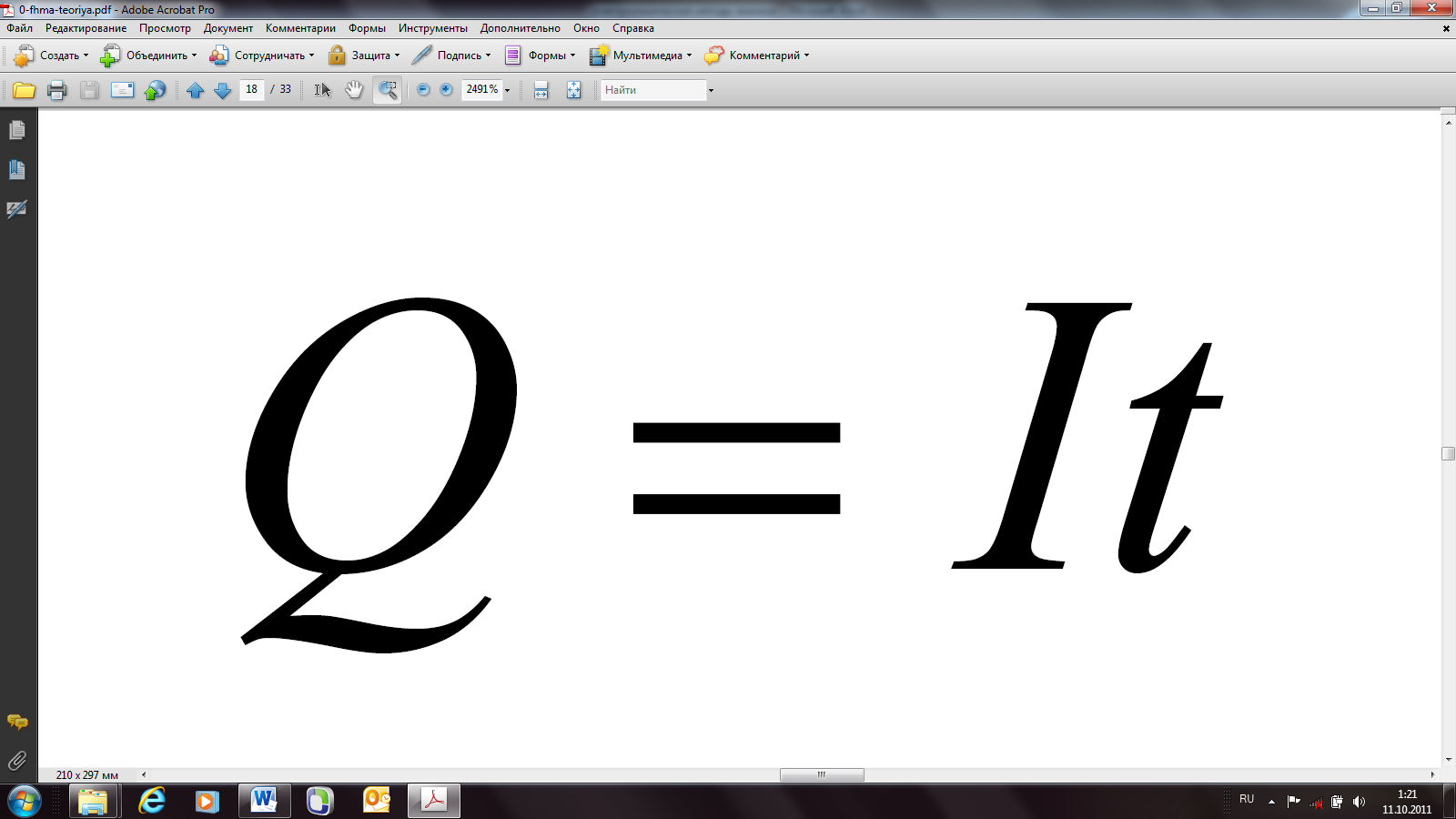
где t – время электролиза, для определения которого пригодны практически все способы установления конечной точки в титриметрии (визуальные – индикаторы, инструментальные – потенциометрия, амперометрия, фотометрия). При силе тока в амперах и времени электролиза в секундах получаем количество электричества в кулонах (пример).
Пример.На кулонометрическое титрование раствора аскорбиновой кислоты иодом, генерируемым из иодида калия током силой 5,00 мА, потребовалось 8 мин 40 с. Рассчитать массу аскорбиновой кислоты в анализируемом растворе. Предложить способ фиксирования конечной точки титрования.
Решение.Количество электричества, затраченное на окисление иодида и, соответственно, аскорбиновой кислоты равно:
Q = Ι·t = 5,00∙10-3∙520 = 2,60 Кл.
Аскорбиновая кислота окисляется иодом до дегидроаскорбиновой кислоты с отдачей двух электронов (С6Н8О6 – 2е → С6Н6О6 + 2Н+), тогда по закону Фарадея:

Конечная точка титрования определяется по появлению избытка иода в растворе. Следовательно, фиксировать ее можно визуально с помощью крахмала, добавленного в анализируемый раствор (появление синей окраски), амперометрически с ртутным капающим или платиновым микроэлектродом по появлению предельного тока иода, потенциометрически по резкому увеличению потенциала платинового электрода.
Вольтамперометрия
Вольтамперометрический метод анализа основан на использовании явления поляризации микроэлектрода, получении и интерпретации вольтамперных (поляризационных) кривых, отражающих зависимость силы тока от приложенного напряжения. Вольтамперная кривая (вольтамперограмма) позволяет одновременно получить качественную и количественную информацию о веществах, восстанавливающихся или окисляющихся на микроэлектроде (деполяризаторах), а также о характере электродного процесса. Современная вольтамперометрия – высокочувствительный и экспрессный метод определения веществ, пригодный для анализа различных объектов неорганической и органической природы, в том числе и фармацевтических препаратов. Минимально определяемая концентрация в вольтамперометрии достигает значений 10-8 моль/л при погрешности метода менее 5%. Вольтамперометрия при оптимальных условиях эксперимента позволяет в анализируемом растворе определять несколько компонентов одновременно.
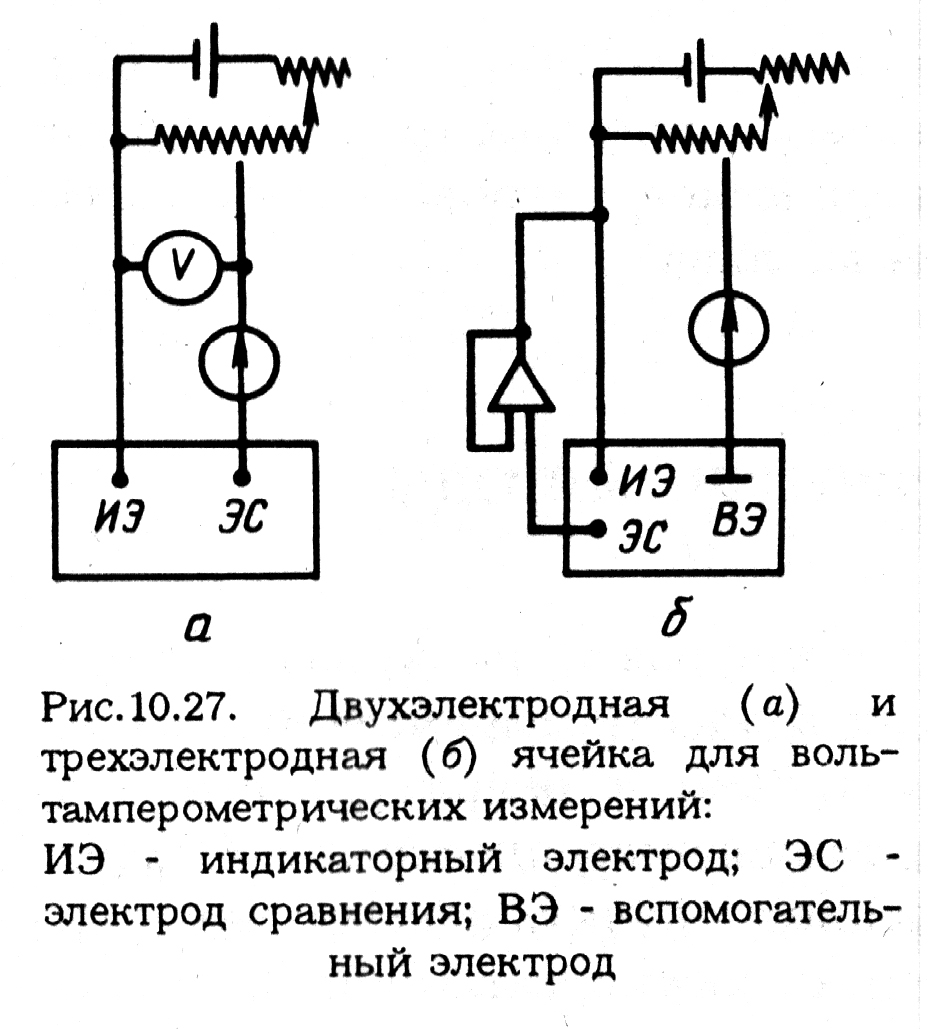
В вольтамперометрии используют два электрода – рабочий поляризуемый электрод с малой поверхностью (индикаторный микроэлектрод) и вспомогательный неполяризуемый электрод с большой поверхностью (электрод сравнения). Рабочими электродами служат микроэлектроды из ртути (ртутный капающий электрод, РКЭ), платины (ПЭ) и токопроводящих углеродных материалов (графит, стеклоуглерод).
| Двухэлектродная (а) и трехэлектродная (б) ячейка для вольтамперометрических измерений |
|
| ИЭ – индикаторный электрод; ЭС – электрод сравнения; ВЭ – вспомогательный электрод |
При прохождении постоянного тока через электролитическую ячейку процесс характеризуется соотношением (закон Ома для раствора электролита):
Е = Ea – Eк + IR
Где Е – приложенное внешнее напряжение; Еа – потенциал анода; Ек – потенциал катода; I – ток в цепи; R – внутреннее сопротивление электролитической ячейки.
При вольтамперометрических измерениях анализируемый раствор содержит индифферентный (фоновый) электролит большой концентрации (в 100 раз и более превышающей концентрацию определяемого вещества – сопротивление раствора мало), а ток в вольтамперометрии не превышает 10-5 А, поэтому падением напряжения в ячейке IR можно пренебречь.
Поскольку в вольтамперометрии один из электродов (вспомогательный) не поляризуется и для него потенциал остается постоянным (его можно принять равным нулю), подаваемое на ячейку напряжение проявляется в изменении потенциала только рабочего электрода и тогда Е = Ea для рабочего микроанода (анодная поляризация) и Е = -Eк для рабочего микрокатода (катодная поляризация). Таким образом, регистрируемая вольтамперная кривая отражает электрохимический процесс, происходящий только на рабочем электроде. Если в растворе присутствуют вещества, способные электрохимически восстанавливаться или окислятся, то при наложении на ячейку линейно изменяющегося напряжения вольтамперограмма имеет форму волны 1 (в отсутствии электрохимической реакции зависимость тока от напряжения линейна 2 в соответствии с законом Ома):
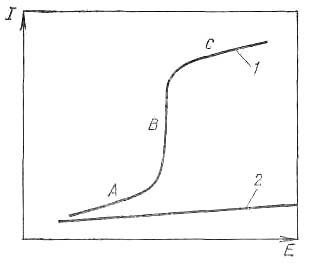
Раздел вольтамперометрии, в котором рабочим микроэлектродом служит РКЭ называют полярографией, в честь чешского электрохимика Я.Гейровского, предложившего этот метод в 1922 году. Вольтамперограммы, полученные в ячейке с ртутным капающим электродом, называют полярограммами.
Для регистрации классических полярограмм ячейку с РКЭ (рабочий электрод) и насыщенным каломельным электродом (вспомогательный электрод, электрод сравнения) присоединяют к источнику постоянного напряжения и изменяют потенциал со скоростью 2-5 мВ/с.
| Простейшая полярографическая ячейка |
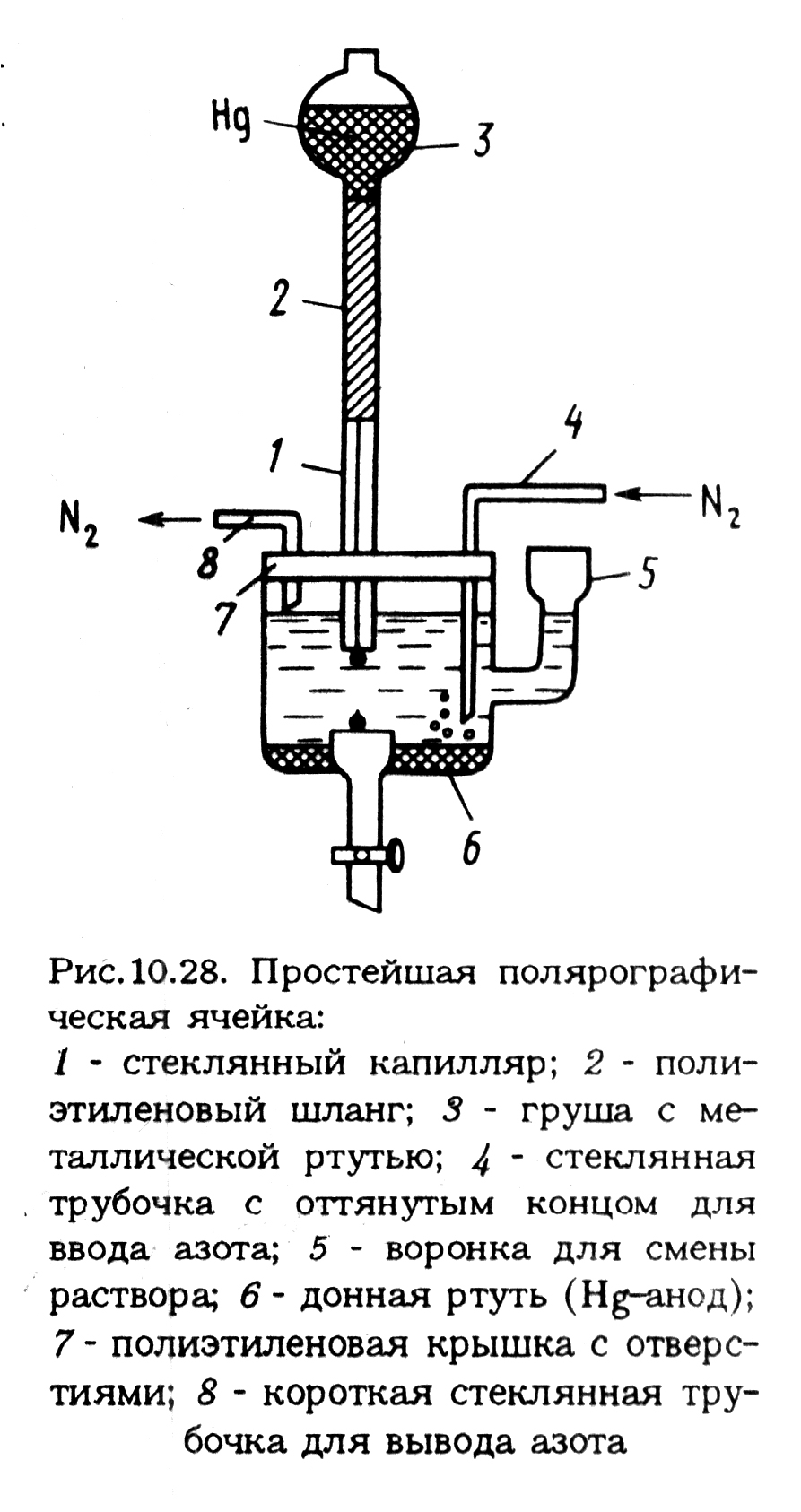
|
| 1 – стеклянный капилляр; 2 – полиэтиленовый шланг; 3 – груша с металлической ртутью; 4 – стеклянная трубочка с оттянутым концом для ввода азота; 5 – воронка для смены раствора; 6 – донная ртуть (Hg-анод); 7 – полиэтиленовая крышка с отверстиями; 8 – короткая стеклянная трубочка для вывода азота |
Ртутный капающий электрод является практически идеально поляризуемым в широком диапазоне потенциалов, ограниченном в анодной области электродными реакциями окисления ртути (+0,4 В), а в катодной реакциями восстановления ионов водорода (от -1 до -1,5 Вв зависимости от кислотности среды) или катионов фона (от -2 В для катионов щелочных металлов до -2,5 В для R4N+). Это позволяет изучать и определять на РКЭ вещества, восстанавливающиеся при очень высоких отрицательных потенциалах, что невозможно на электродах из других материалов. Следует отметить, что здесь и далее значения потенциалов приведены относительно насыщенного каломельного электрода и при необходимости могут быть пересчитаны по отношению к другому электроду сравнения, например, насыщенному хлорсеребряному.
Перед регистрацией полярограммы на РКЭ необходимо удалить растворенный кислород, поскольку он электроактивен в отрицательной области потенциалов, давая две волны восстановления при -0,2 и -0,9 В. Сделать это можно, насыщая раствор инертным газом (азот, аргон, гелий). Из щелочных растворов кислород удаляют с помощью сульфита натрия (O2 + 2Na2SO3 → 2Na2SO4).
Классическая полярограмма (полярографическая волна) в идеализированном виде представлена ниже:
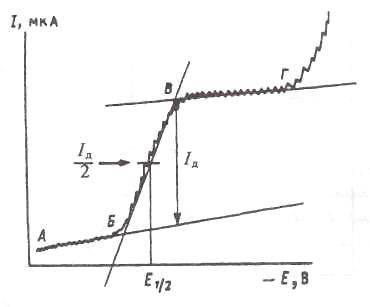
Основными характеристиками полярографической волны являются величина диффузионного тока (Iд, мкА), потенциал полуволны (Е1/2, В) – потенциал, при котором ток равен половине диффузионного, и наклон восходящего участка (0,059/n – крутизна электродной функции). Эти параметры позволяют использовать полярографию как метод анализа (сила тока пропорциональна концентрации) и исследования (потенциал полуволны и электродная функция зависят от природы вещества).
На начальном участке полярографической волны (А-Б) ток с изменением потенциала возрастает очень медленно – это так называемый остаточный ток (Iост). Основной вклад в остаточный ток вносит формирование двойного электрического слоя (ток заряжения), который невозможно исключить и величина которого возрастает с увеличением потенциала. Вторым слагаемым остаточного тока является ток, обусловленный электроактивными примесями, который можно уменьшить применяя чистые реактивы и воду.
При достижении точки Б (потенциал выделения – при восстановлении на катоде потенциал выделения называют потенциалом восстановления Евос, при окислении на аноде – потенциалом окисления Еок) на электроде начинается электрохимическая реакция, в которую вступает электроактивное вещество (деполяризатор), в результате чего ток резко возрастает (участок Б-В) до некоторого предельного значения, оставаясь затем практически постоянным (участок В-Г). Ток, соответствующий этому участку называют предельным током (Iпр), а разность между предельным и остаточным током составляет диффузионный ток (Iд = Iпр – Iост). На участке В-Г при увеличении потенциала предельный и остаточный токи незначительно возрастают, а значение диффузионного тока остается постоянным. Подъем тока в точке Г обусловлен новой электрохимической реакцией (например, восстановлением катионов фонового электролита).
Диффузионный ток получил свое название вследствие того, что в данной области потенциалов в результате электрохимической реакции в приэлектродном слое наблюдается практически полное отсутствие деполяризатора и его обогащение веществом происходит за счет диффузии деполяризатора из глубины раствора, где его концентрация остается постоянной. Поскольку скорость диффузии в данных конкретных условиях остается постоянной, то и диффузионный ток сохраняет постоянство своего значения.
Зависимость величины диффузионного тока от концентрации деполяризатора для р.к.э. выражается уравнением Ильковича:
Id = 605nD1/2m2/3t1/6c
где D – коэффициент диффузии электроактивного иона; n – число электронов, участвующих в реакции; m2/3t1/6 – характеристика капилляра, из которого вытекает ртуть; с - концентрация определяемого вещества (деполяризатора).
При работе с одним и тем же капилляром и деполяризатором значение 605nD1/2m2/3t1/6 = const, поэтому между высотой волны и концентрацией вещества имеется линейная зависимость
Id =КС
На этой линейной зависимости основан количественный полярографический анализ. Взаимосвязь между потенциалом электрода и возникающим током описывается уравнением полярографической волны (уравнение Ильковича-Гейровского):
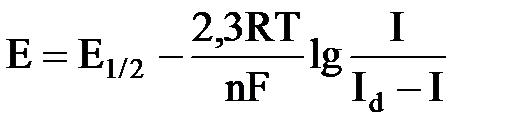
где Е и I – соответственно потенциал и величина тока для данной точки полярографической кривой; Id - величина диффузионного тока; Е1/2 – потенциал полуволны.
Е1/2 - это потенциал, при котором достигается величина тока, равная половине Id. Он не зависит от концентрации деполяризатора. Е1/2 очень близки к нормальному редокс-потенциалу системы (Ео), то есть является качественной характеристикой, определяющейся только природой восстанавливающихся ионов и по которым можно установить качественный состав анализируемого раствора.
Полярограмма (вольтамперограмма) содержит ценную аналитическую информацию – потенциал полуволны Е1/2 является качественной характеристикой деполяризатора (качественный аналитический сигнал), в то время как диффузионный ток Iд линейно связан с концентрацией определяемого вещества в объёме анализируемого раствора (количественный аналитический сигнал) – Iд = KС.
Величина Е1/2 может быть рассчитана из уравнения полярографической волны или определена графически:
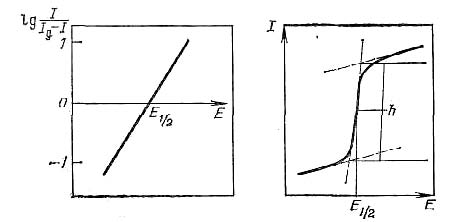
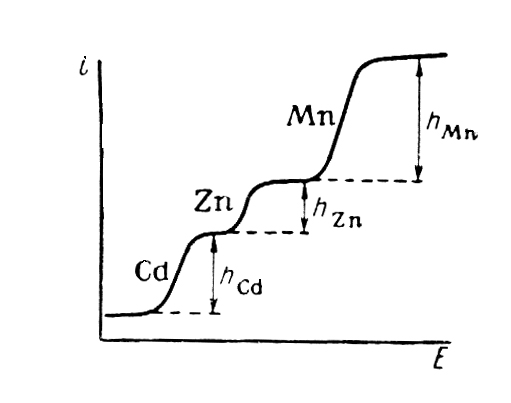
Найденное значение Е1/2 с учетом использованного фонового электролита позволяет на основании табличных данных идентифицировать деполяризатор. Если в анализируемом растворе находится несколько веществ, потенциалы полуволн которых различаются более чем на 0,2 В, то на полярограмме будет не одна волна, а несколько – по числу электроактивных частиц. При этом следует иметь в виду, что восстановление (окисление) многозарядных частиц может происходить ступенчато, давая несколько волн.
| Полярографический спектр |
|
Для исключения перемещения вещества к электроду за счет тепловой и механической конвекции (перемешивания) измерение осуществляется в термостатированном растворе и в отсутствии перемешивания. Устранению электростатического притяжения деполяризатора полем электрода (миграции) способствует большой избыток электронеактивного фонового электролита, ионы которого экранируют заряд электрода, уменьшая движущую силу миграции практически до нуля.
При использовании ртутного капающего электрода на полярограмме наблюдаются осцилляции тока (его периодическое небольшое увеличение и уменьшение). Каждая такая осцилляция соответствует возникновению, росту и отрыву капли ртути от капилляра микроэлектрода. В полярографах предусмотрены устройства для устранения осцилляций.
Полярограммы могут быть искажены за счет полярографических максимумов – резкого возрастания тока выше его предельного значения с последующим спадом:
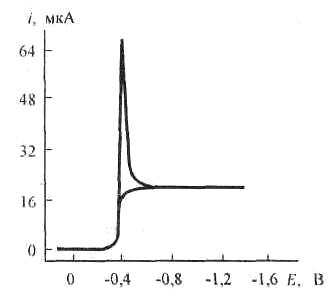
Появление максимумов обусловлено перемешиванием раствора в результате движения поверхности капли ртути из-за неравномерного распределения заряда, а, соответственно, и поверхностного натяжения (максимумы I рода), а также появлений завихрений при вытекании ртути из капилляра (максимумы II рода). Максимумы искажают полярограмму и затрудняют еѐ расшифровку. Для удаления максимумов I рода вводят поверхностно-активное вещество (например, агар-агар, желатин, камфару, фуксин, синтетические ПАВ), которое, адсорбируясь на поверхности ртутной капли, выравнивает поверхностное натяжение и устраняет движение поверхностных слоѐв ртути. Для удаления максимумов II рода достаточно уменьшить давление ртути в капилляре, снизив высоту ртутного столба.
Вольтамперометрия с твердыми рабочими электродами отличается от полярографии с использованием РКЭ другим диапазоном поляризации микроэлектрода. Как было показано выше, ртутный капающий электрод вследствие высокого перенапряжения водорода на нём можно использовать в области высоких отрицательных потенциалов, но из-за анодного растворения ртути при +0,4 В он не может быть применѐн для исследований в области положительных потенциалов. На графите и платине разряд ионов водорода протекает значительно легче, поэтому область их поляризации ограничена значительно более низкими отрицательными потенциалами (-0,4 и -0,1 В соответственно). В то же время в области анодных потенциалов платиновый и графитовый электроды пригодны до потенциала +1,4 В (далее начинается электрохимическая реакция окисления кислорода воды 2Н2О – 4е → О2 + 4Н+), что делает их пригодными для исследований в диапазоне положительных потенциалов.
В отличие от РКЭ во время регистрации вольтамперограммы поверхность твердого микроэлектрода не возобновляется и легко загрязняется продуктами электродной реакции, что приводит к понижению воспроизводимости и точности результатов, поэтому перед регистрацией каждой вольтамперограммы следует проводить очистку поверхности микроэлектрода.
Стационарные твердые электроды не нашли широкого применения в вольтамперометрии из-за медленного установления предельного тока, что приводит к искажению формы вольтамперограммы, однако, на вращающихся микроэлектродах в приэлектродном слое возникают условия для стационарной диффузии, поэтому сила тока устанавливается быстро и вольтамперограмма имеет ту же форму, что и в случае РКЭ.
Величина предельного диффузионного тока на вращающемся дисковом электроде (не зависимо от материала) описывается уравнением конвективной диффузии (Левича):
Id = 0.62nFSD2/3w1/2n-1/6c
где n - число электронов, участвующих в электродном процессе;
F – число Фарадея (96500 кулонов);
S - площадь электрода;
D – коэффициент диффузии деполяризатора;
w - угловая скорость вращения электрода;
n - кинематическая вязкость исследуемого раствора;
с - концентрация деполяризатора, моль/л.
При затруднениях в расшифровке полярограмм применяют метод «свидетеля» – после регистрации полярограммы анализируемого раствора, к нему в электролитическую ячейку поочередно добавляют стандартные растворы предполагаемых соединений. Если предположение было верным, то увеличивается высота волны соответствующего вещества, при неверном предположении появится дополнительная волна при другом потенциале.
Определить концентрацию деполяризатора в анализируемом растворе можно методами градуировочного графика, методом стандарта (сравнения) и методом добавок. При этом во всех случаях следует использовать стандартные растворы, состав которых максимально приближен к составу анализируемого раствора, а условия регистрации полярограмм должны быть одинаковы. Методы применимы в интервале концентраций, где строго соблюдается прямо пропорциональная зависимость диффузионного тока от концентрации деполяризатора. На практике при количественных определениях, как правило, не фиксируют величину диффузионного тока в мкА, а измеряют высоту полярографической волны h, как указано на предыдущем рисунке, которая также является линейной функцией от концентрации h = KC.
По методу градуировочного графика регистрируют полярограммы серии стандартных растворов и строят градуировочный график в координатах h ÷ C (или Iд ÷ С), по которому для найденного значения hx в анализируемом растворе находят концентрацию определяемого вещества в нѐм Сх.
В методе стандарта (сравнения) в одних и тех же условиях записывают полярограммы анализируемого и стандартного растворов определяемого вещества с концентрациями Сх и Сст, тогда:
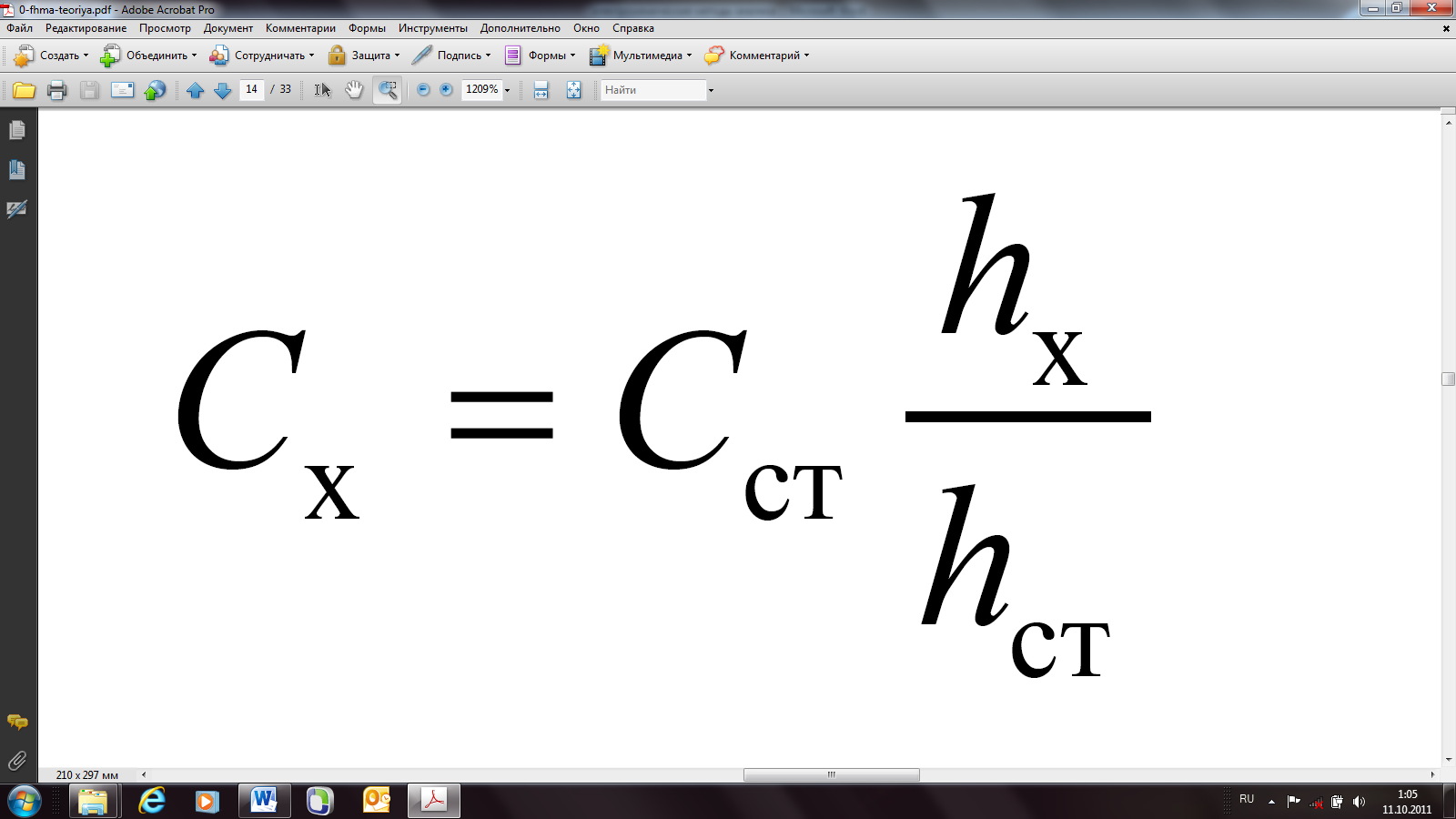
При использовании метода добавок сначала записывают полярограмму анализируемого раствора объемомVx с концентрацией Сх и измеряют высоту волны hx. Затем в электролитическую ячейку к анализируемому раствору добавляют определенный объѐм стандартного раствора определяемого вещества Vд с концентрацией Сд (предпочтительно, чтобы Vx>>Vд и Сх<Сд), записывают полярограмму раствора с концентрацией Сх+д и из-меряют высоту полученной волны hх+д. Несложные преобразования позволяют по этим данным позволяют рассчитать концентрацию определяемого вещества в анализируемом растворе (пример).
Пример. При полярографировании 10,0 мл раствора никотинамида получена волна высотой 38 мм. После добавления к этому раствору 1,50 мл стандартного раствора, содержащего 2,00 мг/мл никотинамида, волна увеличилась до 80,5 мм. Рассчитать содержание препарата (мг/мл) в анализируемом растворе.
Решение. Высота волны никотинамида в анализируемом растворе hx в соответствии с уравнением Ильковича равна:
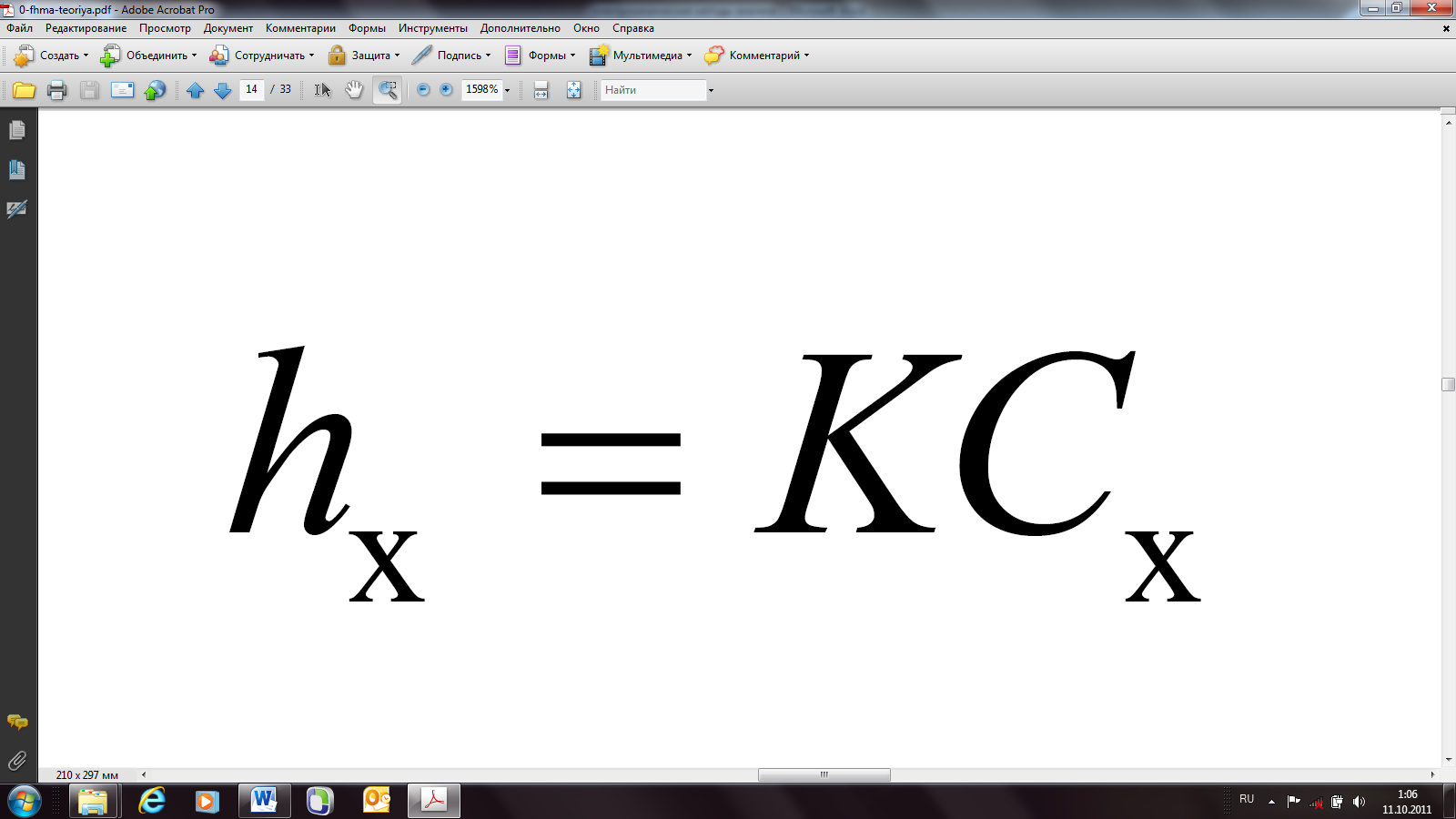
а после добавки стандартного раствора (hх+д):

Если первое уравнение почленно разделить на второе, то получим:

Решая уравнение относительно Сх и подставив значения величин из условия задачи:

Дата добавления: 2015-11-26; просмотров: 5504;