Синтез жирных кислот
Синтез жирных кислот происходит в основном в печени, в меньшей степени – в жировой ткани и лактирующей молочной железе. Гликолиз и последующее окислительное декарбоксилирование пирувата способствуют увеличению концентрации ацетил-КоА в матриксе митохондрий. Синтез же жирных кислот происходит в цитозоле, куда и должен быть транспортирован субстрат. Для этого в матриксе митохондрий ацетил-КоА конденсируется со ЩУК с образованием цитрата. Затем транслоказа переносит цитрат в цитоплазму. Это происходит только при увеличении количества цитрата в митохондриях, когда изоцитратдегидрогеназа и a-кетоглутаратдегидрогеназа ингибированы высокими концентрациями НАДН и АТФ. Такая ситуация создается в абсорбтивном периоде, когда клетка печени получает достаточное количество источников энергии. В цитоплазме цитрат расщепляется до ЩУК и ацетил-КоА. Последний служит исходным субстратом для синтеза жирных кислот, а ЩУК под действием малатдегидрогеназы превращается в малат, который при участии малик-фермента образует пируват. Пируват транспортируется обратно в матрикс митохондрий.
Первая реакция синтеза жирных кислот – превращение ацетил-КоА в малонил-КоА, осуществляемое ацетил-КоА-карбоксилазой, определяет скорость всех последующих реакций синтеза жирных кислот.
Далее синтез жирных кислот продолжается на мультиферментном комплексе – синтазе жирных кислот. Этот фермент состоит из 2 идентичных протомеров, каждый из которых имеет доменное строение и, соответственно, 7 центров, обладающих разными каталитическими активностями (ацетилтрансацилаза, малонилтрансацилаза кетоацилсинтаза, кетоацилредуктаза, гидратаза, еноил-редуктаза, тиоэстераза) и ацилпереносящий белок (АПБ). АПБ не является ферментом, его функция связана только с переносом ацильных радикалов. В процессе синтеза важную роль играют SH-группы. Одна из них принадлежит 4-фосфопантетеину, входящему в состав АПБ, вторая – цистеину кетоацилсинтазы. Протомеры синтазы жирных кислот расположены «голова к хвосту». Несмотря на то, что каждый мономер содержит все активные центры, функционально активен комплекс из двух протомеров. Поэтому реально синтезируются одновременно 2 жирных кислоты (в схемах для упрощения изображают синтез только одной молекулы).
Этот комплекс последовательно удлиняет радикал жирной кислоты на 2 углеродных атома, донором которых служит малонил-КоА. Циклы реакций повторяются до тех пор, пока не образуется радикал пальмитиновой кислоты, который под действием тиоэстеразного центра гидролитически отделяется от ферментного комплекса, превращаясь в свободную пальмитиновую кислоту. В каждом цикле биосинтеза пальмитиновой кислоты проходят 2 реакции восстановления, донором водорода в которых служит НАДФН.
Регуляция синтеза жирных кислот. Регуляторный фермент синтеза жирных кислот – ацетил-КоА-карбоксилаза. Его активность регулируется двумя способами.
1. Ассоциация/диссоциация комплексов субъединиц. В неактивной форме ацетил-КоА-карбоксилаза представляет собой отдельные комплексы, каждый из которых состоит из 4 субъединиц. Активатор фермента – цитрат – стимулирует объединение комплексов, ингибитор – пальмитоил-КоА – вызывает их диссоциацию.
2. Фосфорилирование/дефосфорилирование ацетил-КоА-карбоксилазы. В постабсорбтивном состоянии или при физической работе глюкагон или адреналин через аденилатциклазную систему активируют протеинкиназу А и стимулируют фосфорилирование субъединиц ацетил-КоА-карбоксилазы. Фосфорилированный фермент неактивен, синтез жирных кислот останавливается. В абсорбтивный период инсулин активирует фосфатазу, и ацетил-КоА-карбоксилаза переходит в дефосфорилированное состояние. Затем под действием цитрата происходит полимеризация протомеров фермента, и он становится активным.
Ещё одним способом усиления синтеза жирных кислот является индукция синтеза ферментов этого метаболического пути. Такое происходит при длительном потреблении богатой углеводами и бедной жирами пищи, когда инсулин стимулирует индукцию синтеза ацетил-КоА-карбоксилазы, синтазы жирных кислот, цитратлиазы и изоцитратдегидрогеназы.
Из пальмитиновой кислоты могут синтезироваться более длинные, а также ненасыщенные жирные кислоты. Удлинение пальмитиновой кислоты может происходить:
а) в митохондриях за счет присоединения ацетил-КоА по пути, обратному b-окислению, с использованием НАДФН, а не ФАДН2;
б) в микросомах за счет малонил-КоА и НАДФН. Процесс напоминает функционирование синтазного комплекса в цитозоле, только промежуточные продукты не связываются с АПБ.
Введение двойных связей в структуру жирных кислот происходит также в микросомах с помощью оксидаз, при этом используются НАДФН и О2.
ГЛАВА 21
ОБМЕН СЛОЖНЫХ ЛИПИДОВ
К сложным липидам относят такие соединения, которые, помимо липидного, содержат и нелипидный компонент (белок, углевод или фосфат). Соответственно существуют протеолипиды, гликолипиды и фосфолипиды. В отличие от простых липидов, используемых в качестве энергетического материала, сложные липиды выполняют пластические функции и используются главным образом как структурные компоненты биологических мембран. Протеолипиды являются структурными компонентами в миелиновых оболочках нервных клеток, в синаптических мембранах и внутренних мембранах митохондрий. Гликолипиды участвуют в функционировании мембран: вовлечены в процессы рецепции, участвуют в контроле и регуляции межклеточных контактов. Обладают высокой тканевой специфичностью и выступают в роли антигенов клеточной поверхности. Фосфолипиды (ФЛ) играют важную роль в структуре и функционировании клеточных мембран, активации мембранных и лизосомальных ферментов, в проведении нервных импульсов, свертывании крови, иммунологических реакциях, процессах клеточной пролиферации и регенерации тканей, в переносе электронов в ЦТД.
Образование ФЛ наиболее интенсивно происходит в печени, стенке кишечника, семенниках, яичниках и молочной железе. Синтез ФЛ, содержащих холин и этаноламин начинается с активации азотистых оснований при участии АТФ и соответствующих киназ. При синтезе фосфатидилинозитола на первом этапе происходит взаимодействие фосфатидной кислоты с ЦТФ, ведущее к образованию цитидиндифосфатдиацилглицерола, который реагирует с инозитолом, образуя фосфатидилинозитол.
Помимо путей синтеза индивидуальных ФЛ, имеются пути их взаимопревращений, целесообразность которых, очевидно, связана с необходимостью обеспечения тканей требуемым ФЛ в нужный момент.
Для синтеза фосфатидилхолинов, и в меньшей степени – сфингомиелинов, нужен холин или метионин, потребность в которых в значительной степени покрывается за счет пищевых источников. При длительном недостатке в пище холина и метионина наблюдается развитие жировой инфильтрации печени, при которой содержание липидов, главным образом ТАГ, может достигать в расчете на сухую массу ткани 45 %, против 7-14 % в норме. Механизм развития жировой инфильтрации печени связан с недостаточным синтезом фосфатидилхолинов и сфингомиелинов, необходимых для формирования в этом органе ЛП. На образование последних, наряду с ФЛ, используются значительные количества ТАГ и холестерола. Сформированные в печени ЛП, в частности богатые триацилглицеролами ЛПОНП, поступают в кровяное русло. Следовательно, образование ЛП можно рассматривать как важнейший путь утилизации печеночных липидов. Поэтому недостаточный синтез в печени содержащих холин ФЛ нарушает образование ЛП и ведет к накоплению в этом органе ТАГ и ХС. По этой причине холин, метионин, а также фосфатидилхолин относятся к группе липотропных веществ, прием которых с пищей предотвращает развитие жировой инфильтрации печени.
Распад фосфолипидов может происходить при участии нескольких ферментов, каждый из которых катализирует гидролитический разрыв строго определеннной связи. Гидролиз некоторых ФЛ под действием фосфолипаз имеет значение не только как путь катаболизма, но и как путь образования эйкозаноидов. Кроме того, фосфолипазы А1 и А2 участвуют в изменении состава жирных кислот в ФЛ, например при синтезе в эмбриональном периоде дипальмитоилфосфатидилхолина – компонента сурфактанта.
Для образования гликолипидов и сфингомиелина (сфинголипидов) вначале требуется синтез самого сфингозина. Это происходит путем конденсации пальмитоил-КоА с серином при участии пиридоксальфосфата (ПАЛФ) и ионов марганца
(Рис. 21.1.).
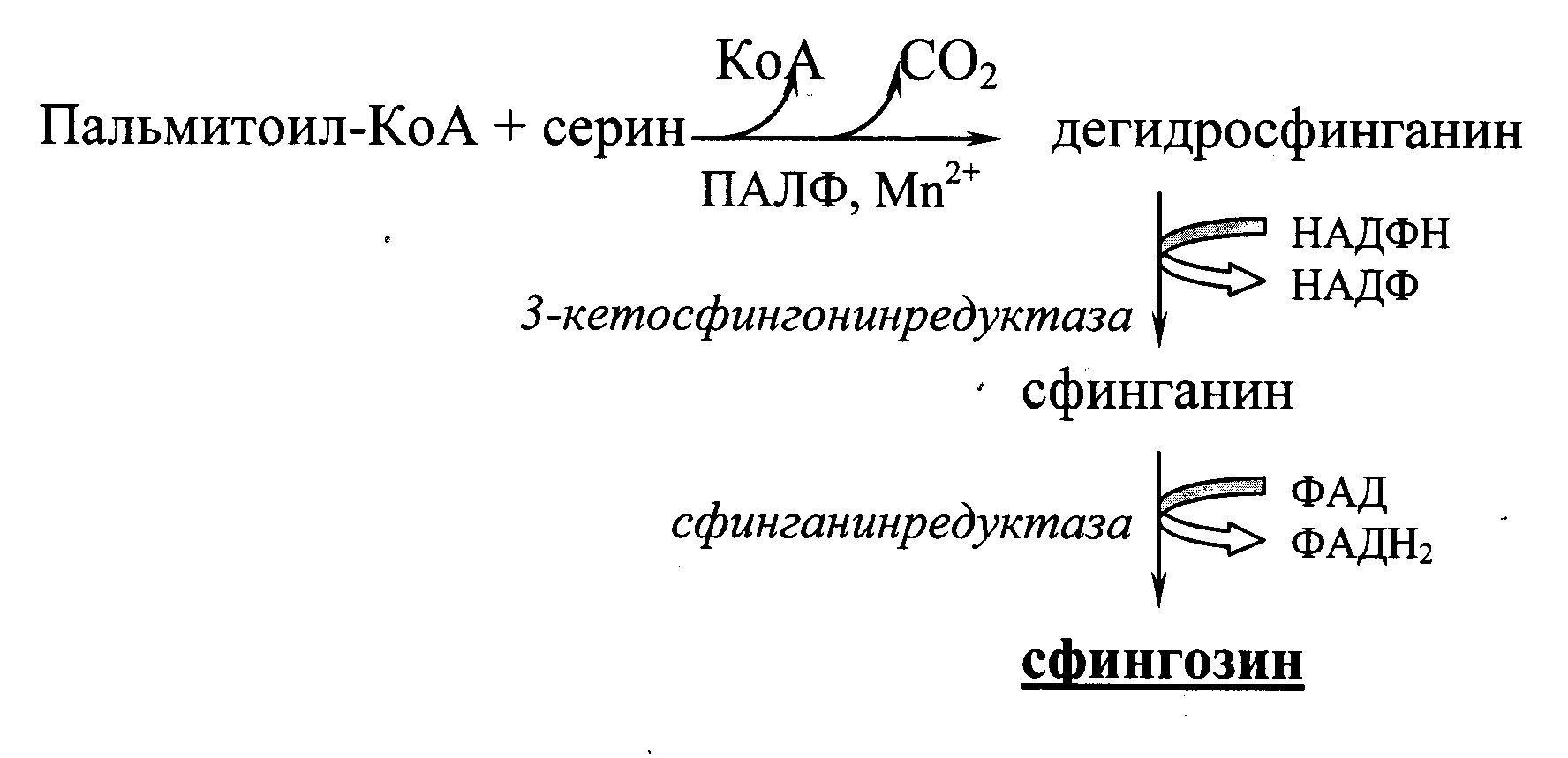
Рис. 21.1. Схема образования сфингозина
Сфингозин подвергается ацилированию (присоединение остатка жирной кислоты), в результате образуется церамид,из которого могут синтезироваться цереброзиды, ганглиозиды, сульфатиды и сфингомиелин (Рис.21.2.).
Катаболизм сфингомиелинов и гликолипидов происходит в лизосомах. Исключительно важный аспект этого процесса заключается в существовании более десяти специфических лизосомных болезней накопления – сфинголипидозов. Сфинголипидозы обычно являются причиной умственной отсталости и ведут к смерти в раннем возрасте, так как происходит поражение клеток нервной ткани, где сконцентрированы гликолипиды.
В распаде сфингомиелинов (Рис.21.2.) участвует сфингомиелиназа, отщепляющая фосфохолин. Генетический дефект сфингомиелиназы – причина болезни Нимана-Пика. Дети с таким дефектом погибают в раннем возрасте. Симптомы болезни: накопление сфингомиелина в лизосомах, умственная отсталость, гепатоспленомегалия.
Сложные молекулы гликолипидов расщепляются в результате последовательных реакций гидролиза до глюкозы, галактозы, церамида и других метаболитов. Генетические дефекты любого из ферментов, обеспечивающих катаболизм этого класса липидов,
ведут к развитию заболеваний, среди которых можно назвать:
· болезнь Гоше – следствие дефекта b-глюкозидазы, при которой наблюдаются гепатоспленомегалия и умственная отсталость;
· болезнь Тея-Сакса – следствие дефекта b-гексозаминидазы, для которой характерны умственная отсталость и слепота;
· генерализованный ганглиозидоз, вызываемый снижением активности b-галактозидазы, также ведущий к умственной отсталости.
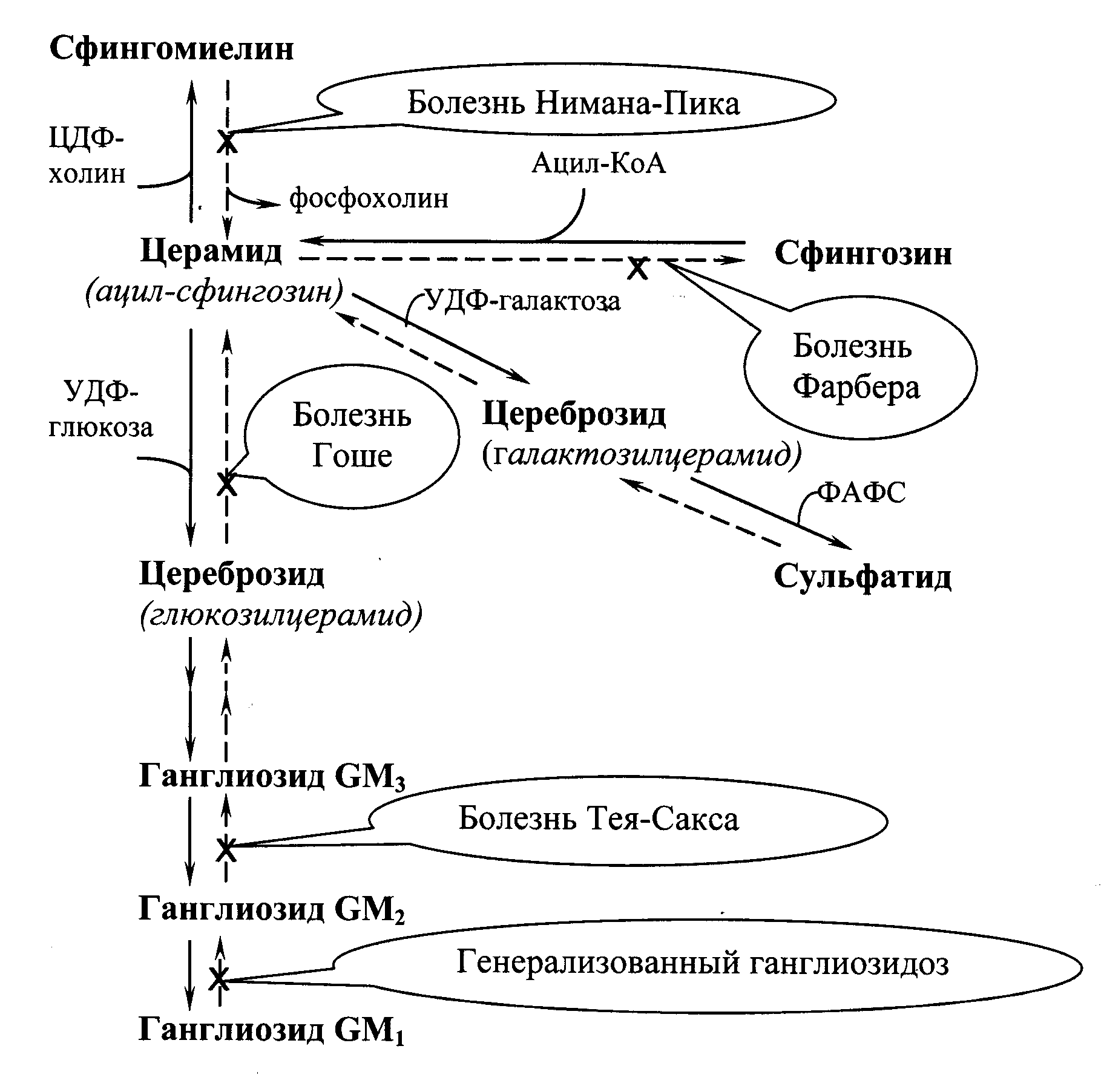
Рис.21.2. Биосинтез ( ) и распад ( ) сфинголипидов с указанием биохимических нарушений при сфинголипидозах.
Расщепление церамида до сфингозина и жирной кислоты осуществляется церамидазой. Генетический дефект этого фермента приводит к развитию болезни Фарбера с летальным исходом в раннем возрасте. При данной патологии в лизосомах накапливается церамид, наблюдается гепатоспленомегалия, умственная отсталость и поражения суставов.
ГЛАВА 22
МЕТАБОЛИЗМ ХОЛЕСТЕРОЛА.
БИОХИМИЯ АТЕРОСКЛЕРОЗА
Холестерол – стероид, характерный только для животных организмов. Основное место его образования в организме человека – печень, где синтезируется 50% холестерола, в тонком кишечнике его образуется 15-20%, остальное количество синтезируется в коже, коре надпочечников и половых железах. Источники формирования фонда холестерола и пути его расходования представлены
на рис 22.1.
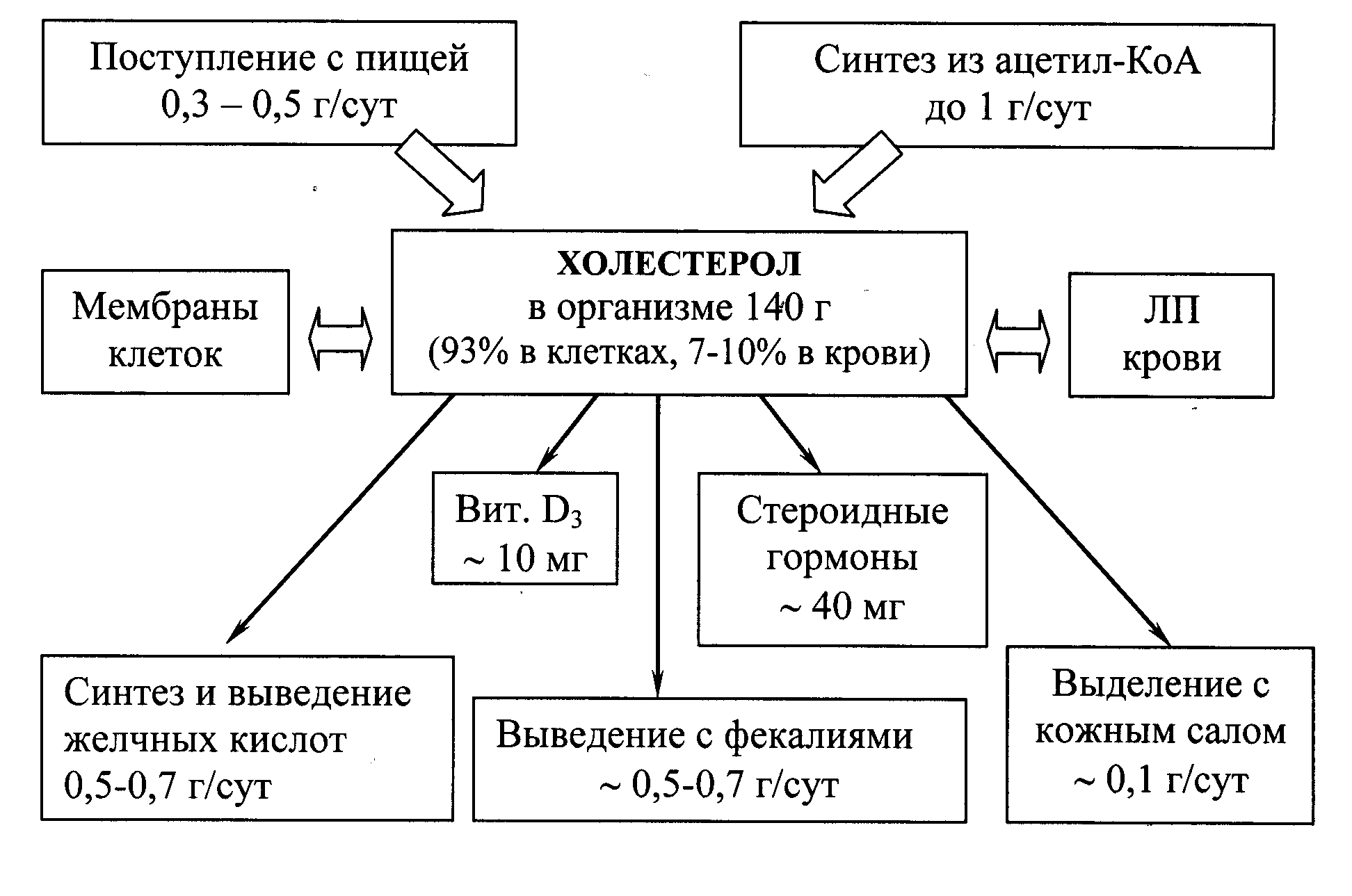
Рис. 22.1. Формирование и распределение фонда холестерола в организме.
Холестерол организма человека (суммарное количество около 140 г) условно можно разделить на три пула:
пул А (~ 30 г),быстрообменивающийся, состоит из ХС кишечной стенки, плазмы крови, печени и других паренхиматозных органов, обновление происходит за 30 сут (1 г/сут);
пул Б(~ 50 г), медленнообменивающийся ХС остальных органов и тканей;
пул В(~ 60 г),очень медленнообменивающийся ХС спинного и головного мозга, соединительной ткани, скорость обновления исчисляется годами.
Синтез холестерола происходит в цитозоле клеток. Это один из самых длинных метаболических путей в организме человека. Он проходит в 3 этапа: первый заканчивается образованием мевалоновой кислоты, второй – образованием сквалена (углеводород линейной структуры, состоящий из 30 углеродных атомов). В ходе третьего этапа сквален превращается в молекулу ланостерола, далее происходит 20 последовательных реакций, превращающих ланостерол в холестерол.
В некоторых тканях гидроксильная группа холестерола этерифицируется с образованием эфиров. Реакция катализируется внутриклеточным ферментом АХАТ (ацилКоА:холестеролацилтрансферазой). Реакция этерификации происходит также в крови в ЛПВП, где находится фермент ЛХАТ (лецитин:холестеролацилтрансфераза). Эфиры холестерола – форма, в которой он транспортируется кровью или депонируется в клетках. В крови около 75% ХС находится в виде эфиров.
Регуляция синтеза холестерола осуществляется путем влияния на активность и количество ключевого фермента процесса – 3-гидрокси-3-метилглутарил-КоА-редуктазы (ГМГ-КоА-редуктазы). Это достигается двумя способами:
1. Фосфорилирование/дефосфорилирование ГМГ-КоА-редуктазы. Инсулин стимулирует дефосфорилирование ГМГ-КоА-редуктазы, переводя её тем самым в активное состояние. Следовательно, в абсорбтивный период синтез ХС увеличивается. В этот период увеличивается и доступность исходного субстрата для синтеза – ацетил-КоА. Глюкагон оказывает противоположное действие: через протеинкиназу А стимулирует фосфорилирование ГМГ-КоА-редуктазы, переводя её в неактивное состояние. В результате синтез ХС в постабсорбтивном периоде и при голодании ингибируется.
2. Ингибирование синтеза ГМГ-КоА-редуктазы. ХС (конечный продукт метаболического пути) снижает скорость транскрипции гена ГМГ-КоА-редуктазы, подавляя таким образом собственный синтез, аналогичный эффект вызывают и жёлчные кислоты.
Транспорт холестерола кровью осуществляется в составе ЛП. ЛП обеспечивают поступление в ткани экзогенного ХС, определяют его потоки между органами и выведение из организма. Экзогенный ХС доставляется в печень в составе остаточных ХМ. Там вместе с синтезированным эндогенным ХС он формирует общий фонд. В гепатоцитах ТАГ и ХС упаковываются в ЛПОНП, и в таком виде секретируются в кровь. В крови ЛПОНП под действием ЛП-липазы, гидролизующей ТАГ до глицерола и жирных кислот, превращаютя сначала в ЛППП, а затем и в ЛПНП, содержащие до 55% ХС и его эфиров. ЛПНП – основная транспортная форма ХС, в которой он доставляется в ткани (70% ХС и его эфиров в крови находится в составе ЛПНП). Из крови ЛПНП поступают в печень (до 75%) и другие ткани, которые имеют на своей поверхности рецепторы ЛПНП.
Если количество ХС, поступающего в клетку, превышает её потребность, то синтез рецепторов ЛПНП подавляется, что уменьшает поток ХС из крови. При снижении концентрации свободного ХС в клетке, наоборот, синтез рецепторов активируется. В регуляции синтеза рецепторов ЛПНП участвуют гормоны: инсулин, трийодтиронин и половые гормоны увеличивают образование рецепторов, а глюкокортикоиды – уменьшают.
В так называемом «обратном транспорте холестерола», т.е. пути, обеспечивающем возвращение ХС в печень, основную роль играют ЛПВП. Они синтезируются в печени в виде незрелых предшественников, которые практически не содержат ХС и ТАГ. В крови предшественники ЛПВП насыщаются ХС, получая его из других ЛП и мембран клеток. В переносе ХС в ЛПВП участвует фермент ЛХАТ, находящийся на их поверхности. Этот фермент присоединяет остаток жирной кислоты от фосфатидилхолина (лецитина) к ХС. В результате образуется гидрофобная молекула эфира холестерола, которая перемещается внутрь ЛПВП. Таким образом, незрезые ЛПВП, обогащаясь ХС, превращаются в ЛПВП3 – зрелые и более крупные по размерам частицы. ЛПВП3 обменивают эфиры холестерола на ТАГ, содержащиеся в ЛПОНП и ЛППП при участии специфического белка, переносящего эфиры холестерола между липопротеинами. При этом ЛПВП3 превращаются в ЛПВП2, размер которых увеличивается за счет накопления ТАГ. ЛПОНП и ЛППП под действием ЛП-липазы превращаются в ЛПНП, которые в основном и доставляют ХС в печень. Небольшая часть ХС доставляется в печень ЛПВП2 и ЛППП.
Синтез жёлчных кислот.В печени из ХС синтезируется 500-700 мг жёлчных кислот в сутки. Их образование включает реакции введения гидроксильных групп при участии гидроксилаз и реакции частичного окисления боковой цепи ХС (Рис. 22.2):
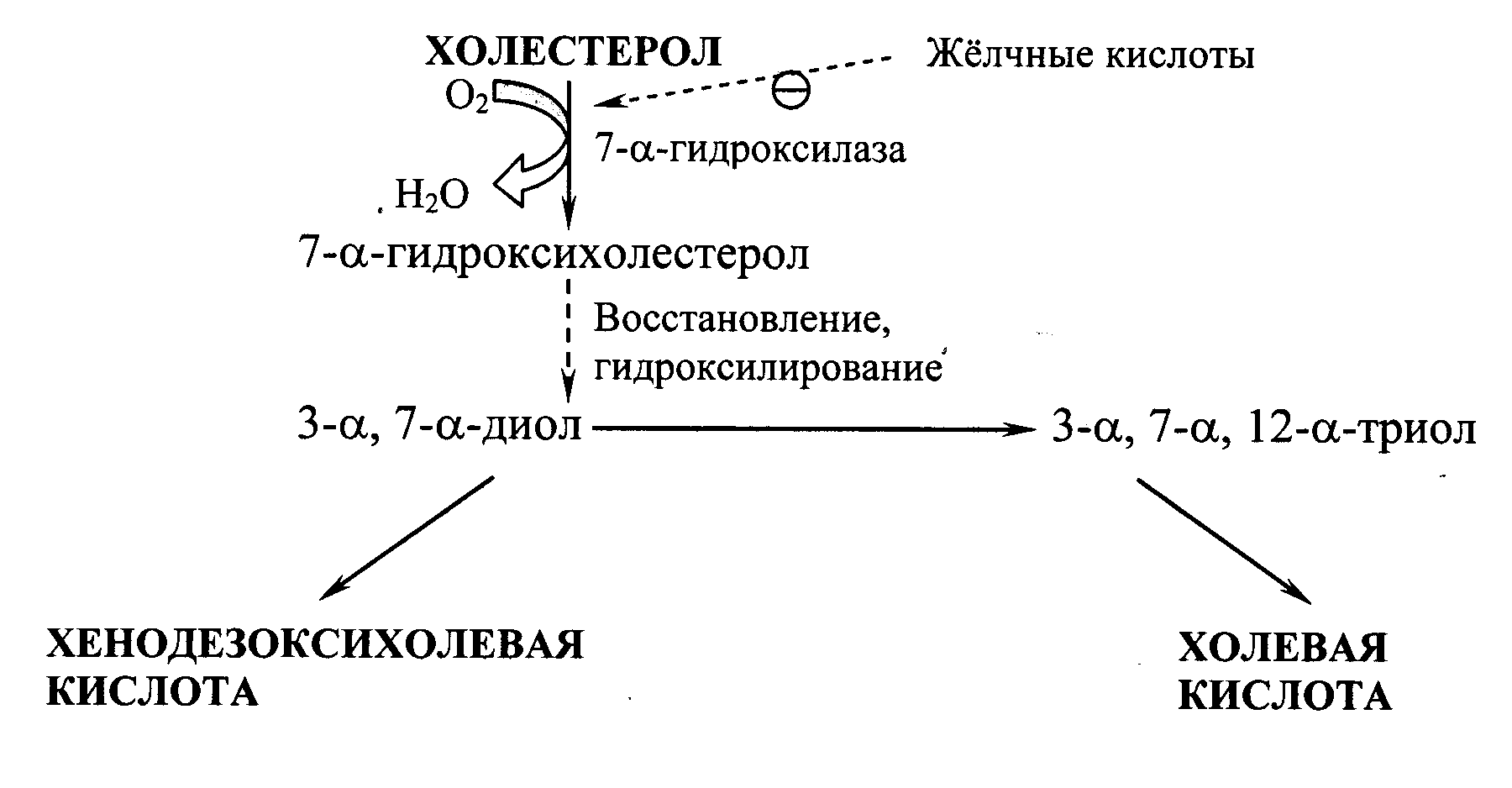
Рис. 22.2. Схема образования жёлчных кислот.
Первая реакция синтеза – образование 7-a-гидроксихолестерола – является регуляторной. Активность фермента, катализирующего эту реакцию, ингибируется конечным продуктом пути – жёлчными кислотами. Еще одним механизмом регуляции является фосфорилирование/дефосфорилирование фермента (активна фосфорилированная форма 7-a-гидроксилазы). Возможна и регуляция путем изменения количества фермента: ХС индуцирует транскрипцию гена 7-a-гидроксилазы, а жёлчные кислоты репрессируют. Тиреоидные гормоны индуцируют синтез 7-a-гидроксилазы, а эстрогены – репрессируют. Такое влияние эстрогенов на синтез жёлчных кислот объясняет, почему желчнокаменная болезнь встречается у женщин в 3-4 раза чаще, чем у мужчин.
Образовавшиеся из ХС холевую и хенодезоксихолевую кислоты называют «первичными жёлчными кислотами». Основная масса этих кислот подвергается коньюгации – присоединению молекул глицина или таурина к карбоксильной группе жёлчной кислоты. Коньюгация начинается с образования активной формы желчных кислот – производных КоА, затем присоединяются таурин или глицин, и в результате образуется 4 варианта коньюгатов: таурохолевая и таурохенодезоксихолевая, гликохолевая и гликохенодезоксихолевая кислоты. Они являются значительно более сильными эмульгаторами, чем исходные жёлчные кислоты. Коньюгатов с глицином образуется в 3 раза больше, чем с таурином, так как количество таурина в организме ограничено. В кишечнике небольшое количество коньюгатов первичных жёлчных кислот под действием ферментов бактерий превращаются во вторичные жёлчные кислоты. Дезоксихолевая кислота, образующаяся из холевой, и литохолевая, образующаяся из дезоксихолевой, хуже растворимы и медленнее всасываются в кишечнике.
Около 95% жёлчных кислот, попавших в кишечник, возвращаются в печень через воротную вену, затем опять секретируются в жёлчь и повторно используются в эмульгировании жиров. Этот путь жёлчных кислот называется энтерогепатической циркуляцией. С фекалиями в основном удаляются вторичные жёлчные кислоты.
Желчнокаменная болезнь(ЖКБ) – патологический процесс, при котором в жёлчном пузыре образуются камни, основу которых составляет ХС.
Выделение ХС в жёлчь должно сопровождаться пропорциональным выделением жёлчных кислот и фосфолипидов, удерживающих гидрофобные молекулы ХС в мицеллярном состоянии. Причинами, приводящими к изменению соотношения жёлчных кислот и ХС в жёлчи являются: пища, богатая ХС, высококалорийное питание, застой жёлчи в жёлчном пузыре, нарушение энтерогепатической циркуляции, нарушения синтеза жёлчных кислот, инфекции жёлчного пузыря.
У большинства больных ЖКБ синтез ХС увеличен, а синтез жёлчных кислот из него замедлен, что приводит к диспропорции количества ХС и жёлчных кислот, секретируемых в жёлчь. В итоге ХС начинает осаждаться в жёлчном пузыре, образуя вязкий осадок, который постепенно затвердевает. Иногда он пропитывается билирубином, белками и солями кальция. Камни могут состоять только из ХС (холестериновые камни) или из смеси ХС, билирубина, белков и кальция. Холестериновые камни обычно белого цвета, а смешанные – коричневые разных оттенков.
В начальной стадии образования камней можно применять в качестве лекарства хенодезоксихолевую кислоту. Попадая в жёлчный пузырь, она постепенно растворяет холестериновые камни, однако это медленный процесс, длящийся несколько месяцев.
Дата добавления: 2015-10-05; просмотров: 1201;