Защита функциональных групп как универсальный способ управления селективностью реакций
Во всех подходах к проблеме селективности, которые мы рассматривали вы-ше, «игра» строилась на вариациях, непосредственно затрагивающих участ-ников основного процесса: изменялись природа субстрата и/или реагента, условия проведения реакции или даже природа самой реакции. Хотя в каж-дом из случаев удавалось обеспечить селективность требуемого превращения, однако подчас этот успех достигался дорогой ценой, поскольку требова-лось «подогнать» к решению той или иной конкретной задачи какой-либо из основных методов синтеза, иначе говоря, используя применявшуюся нами ранее метафору «влезть внутрь черного ящика». На практике во многих слу-чаях. оказывается более выгодным иной подход к проблеме селективности. Поясним его на следующем схематическом примере.
Рассмотрим некий субстрат А—X, для которого хорошо отработан метод его превращения в продукт A-Z. Допустим теперь, что конкретная задача состоит в селективном превращении субстрата Y—А—X, где Z — группа, близкая по свойствам группе X, в продукт Y—A-Z. Можно, конечно попытаться, например, модифицировать основную реакцию так, чтобы она затрагивала только группу X и совершенно не затрагивала группу Y. Однако такой путь может оказаться очень трудоемким, поскольку придется модифицировать уже хорошо отработанный и, возможно, сложный метод, причем не исключено, что для каждого нового Y в системах типа Y"—А—X эту работу придется проделывать заново. К счастью, существует иной принцип решения такого рода задач. Суть гго состоит в том, чтобы временно вывести из игры группу Y и тем самым превратить бифункциональный субстрат Y-A-X в монофункциональный, к которому применим обычный метод трансформации X в Z в его канонической форме. Этого можно добиться использованием некоторых простейших реакций, превращающих функцию Y в группу, инертную в условиях основной реакции и допускающую безболезненный возврат от нее к исходной функции Y на более поздних стадиях синтеза.
Такая маскировка, или защита функций, — прием, чрезвычайно широко используемый в практике органического синтеза. Легко видеть, что при этом снимается проблема селективности основной реакции, но появляется вопрос о селективности постановки защитной группы на функцию Z без затрагивания родственной функции X. Однако в общем случае найти решение этой задачи уже несравненно легче по ряду причин. Во-первых, методы введения защит относятся к категории трансформаций функциональных групп, которые сравнительно просты по химизму и для которых отработаны десятки методов, что делает их применимыми практически для всех мыслимых случаев. Во-вторых, структуру защитной группы можно варьировать в очень широких пределах, поскольку на последующих стадиях она будет удалена, и ее характер не может повлиять на образование последующих продуктов синтетической цепочки*. Благодаря этим обстоятельствам диапазон реакций, которые могут быть использованы для защиты данной функциональной группы, чрезвычайно широк, что надежно обеспечивает требуемую селективность постановки защитной группы. Для иллюстрации применения «защитного подхода» к проблеме селективности рассмотрим восстановление уже знакомой нам модельной трифун-кциональной системы 156 (схема 2.86).
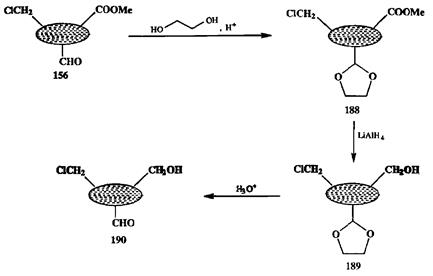 Схема 2.86
Схема 2.86
|
Ранее на этой же системе мы показали, каким образом можно добиться селективного восстановления только формильной группы или формильной икарбометоксильной групп за счет вариаций природы гидридного восстановителя (см. схему 2.73). А как быть, если требуется восстановить селективно только карбометокигруппу? Если учесть, что эта функция по отношению к любому из общепринятых гидридных восстановителей будет менее активна, чем формильная группа, то может показаться, что требуемое превращение вообще невозможно провести с использованием реагентов этого типа. Однако на самом деле ситуацию легко исправить, если защитить карбонильную группу, превратив ее в ацетальную с помощью, например, кислотно-катализируемой реакции с этиленгликолем. Поскольку ацетали устойчивы к действию самых разных нуклеофилов, сложноэфирную группу модифицированного субстрата 188 можно восстанавливать с помощью любого гидридного восстановителя. Получаемый при этом спирт 189 отличается от требуемого продукта 190 лишь наличием ацетильной защиты, но последняя легко удаляется кислотно-катализируемым гидролизом. Таким образом, почти неразрешимая проблема селективного восстановления карбометоксигуппы в присутствии легко восстанавливаемой альдегидной функции легко решается при использовании «защитного подхода».
Разберем теперь более конкретно некоторые методы защиты важнейших функциональных групп, начиная с карбонильной функции.
Упомянутая выше аиетальная зашита в принципе может быть поставлена на любое карбонильное соединение с использованием самых различных спиртов или гликолей, но скорость этой реакции в зависимости от конкретной природы субстрата может различаться на несколько порядков. Это позволяет, в частности, четко дифференцировать альдегидную и кетонную функцию, поскольку первая является более активным электрофилом и существенно легче может быть превращена в ацеталь. Рассмотрим в качестве примера конкретную синтетическую задачу, в которой эффективно был использован именно этот прием.
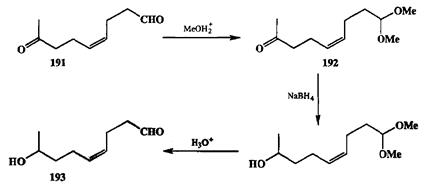 Схема 2.87
Схема 2.87
|
Легко доступный кетоальдегид 191(схема 2.87) является перспективным исходным веществом для синтеза ряда феромонов [26а]. Одна из схем синтеза требовала селективного восстановления кетонного карбонила в этом соединении. В мягких условиях ацетализации (слабая кислота, метанол) затрагивалась только ачьдегидная группа, и монозамещенное производное 192удалось получить с хорошим выходом. Восстановление кетогруппы в этом соединении под действием натрийборгидрида с последующим снятием аце-тальной защиты дало требуемый продукт 193.
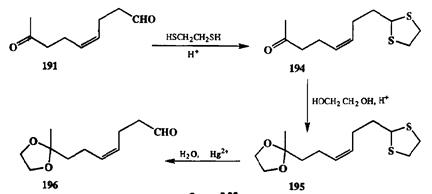 Схема 2.88
Схема 2.88
|
На этом же примере удобно показать, каким образом может быть обеспечена обратная селективность восстановления. Для этой цели сначала защищают альдегидную группу постановкой тиоацетальной защиты (схема 2.88). Поскольку тиоацетали достаточно устойчивы в слабокислых условиях, полученный продукт 194может быть далее превращен в дизащищенное производное 195.Специфической особенностью тиоацеталей является их способность достаточно легко претерпевать сольволиз при обработке солями ртути (или кадмия). Путем такой обработки из продукта 195получают монозамещенное производное 196,в котором на этот раз защищена кетогруппа, а альдегидная группа может быть далее восстановлена или использована в любых других реакциях с нуклеофильными реагентами.
Нередки случаи, когда требуется дифференцировать обычную карбонильную группу и такую же группу, находящуюся в сопряжении с двойной связью. Поскольку наличие такого сопряжения существенно снижает элект-рофильность карбонильного центра, ацетализация в подобного рода полифункциональных системах будет протекать с высокой селективностью, затрагивая лишь изолированную карбонильную функцию. Этот прием, особенно часто применяемый в химии стероидов [26Ь], позволяет на последующих стадиях использовать сохранившуюся в молекуле еноновую группировку в таких превращениях, как, например, присоединение по Михаэлю.
Проблемы, которые возникают при необходимости осуществить селективную защиту гидроксильных групп, удобно рассмотреть на примерах из химии углеводов. Допустим, что нам надо селективно провести реакцию по первичной гидроксильной группе при С-6 а-метил-О-глюкопиранозида (197)(схема 2.89).
Очевидно, что для достижения этой цели необходимо в первую очередь защитить три другие имеющиеся в молекуле гидроксильные функции. Во-можный способ решения этой задачи — синтез триацетата 198.Однако прямое превращение 197в 198трудно осуществить, ибо ацетилирование — малоселективная реакция, протекающая с первичными спиртами быстрее, чем со вторичными. Поэтому приходится прибегнуть к обходному маневру — к синтезу трифенилметильного (тритильного, Тг) эфира 199.Введение три-тильной защиты по первичным гидроксилам осуществляется легче, чем по вторичным, поскольку реакции объемистой тритильной группы очень чувствительны к пространственному экранированию атакуемого центра. Действительно, обработка глюкозида 197тритилхлоридом в пиридине с высоким выходом приводит к монотритиловому эфиру 199.В этом соединении защищен первичный гидроксил, который должен быть свободным в целевом соединении. Это, однако, не должно нас смущать: главное в том, что нам удалось его как-то «пометить», т.е. отличить от других. На следующей стадии нам требуется закрыть все остальные гидроксильные группы, для чего вполне можно воспользоваться стандартной методикой ацетилирования уксусным ангидридом в пиридине. В полученном производном 200имеются два типа защитных групп, резко различающихся по своим свойствам, в частности, по стабильности по отношению к кислотным реагентам. Поэтому превращение этого продукта в целевой триацетат 198может быть осуществлено с высокой селективностью с помощью гидролиза в слабокислой среде.
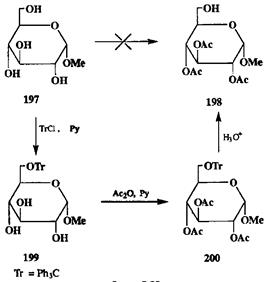 Схема 2.89
Схема 2.89
|
На рассмотренном примере поучительно проследить некоторые общие принципы использования защитных групп. Селективность конечного результата в показанной последовательности превращений достигается, с одной стороны, селективностью введения первой защиты, обусловленной как ее свойствами, так и свойствами защищаемой функции, а с другой — селективностью удаления одной из защит, обусловленной уже только различиями в свойствах этих групп как таковых. Таким образом, селективность введения защиты и селективность ее удаления управляются совершенно различными факторами и поэтому составляют два мощных и независимых способа управления селективностью всего синтеза.
Задача селективной защиты гидроксильной группы возникает чрезвычайно часто в полном синтезе. Именно поэтому для спиртовой функции создана весьма изощренная система защит буквально «на все случаи жизни». Некоторые из наиболее употребимых защит приведены на схеме 2.90. Все показанные производные относятся к числу в общем-то вполне обычных продуктов трансформации гидроксильной группы: это сложные эфиры (201-203),ацетали (204, 205),простые эфиры (206-209)и силиловые эфиры (210, 211)[26c,d]. Получение всех этих производных осуществляется по обшей схеме электро-фильного замещения водорода гидроксильной группы, однако методы введения конкретных защит различаются весьма сильно и охватывают и кислую, и нейтральную, и щелочную области. Легкость протекания реакции постановки той или иной защиты зависит от природы спиртового гидроксила, т. е. от особенностей строения фрагмента, содержащего гидроксильный заместитель. Так, например, относительная реакционоспособность спиртов в таких реакциях может быть представлена рядом: «-AlkOH > в/яо/>-А1ЮН > трет-МкОИ; экваториальный ROH > аксиальный ROH. Используя различия в реакционной способности спиртовых функций, можно достаточно тонко дифференцировать эти группы путем селективного введения подходящих защит.
Диапазон условий, в которых устойчивы защиты спиртовых гидроксилов, охватывает практически всю область, в которой могут проводиться основные реакции, применямые в органическом синтезе (кроме суперкислых сред). В целом для простых эфиров, ацеталей и кеталей характерна высокая устойчивость по отношению к основаниям и нуклеофилам, а также к окислителям и восстановителям; для сложных эфиров — к электрофилам и окислителям и в довольно широком диапазоне к кислотам; для силиловых эфиров — к окислителям и восстановителям и электрофилам некоторых типов. Поэтому для обеспечения сохранности спиртовой группы в условиях практически любой реакции, протекающей с участием других имеющихся функций, всегда можно подобрать какую-либо защиту из имеющегося богатого набора вариантов.
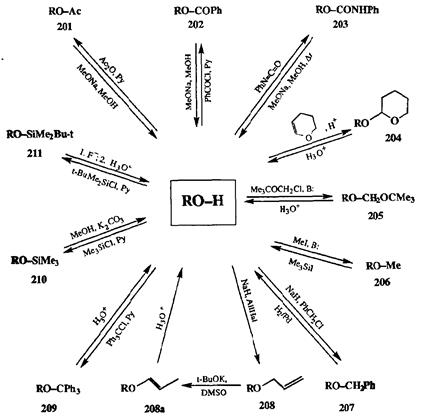 Схема 2.90
Схема 2.90
|
Условия снятия перечисленных защит также весьма разнообразны: это кислый или щелочной сольволиз, каталитический гидрогенолиз, восстановление комплексными гидридами или щелочными металлами в жидком аммиаке и расщепление под действием таких специфических реагентов, как, например, несольватированный фторид-ион (для силиловых производных) или триметилиодсилан (для метиловых эфиров, стабильных к большинству остальных реагентов). В пределах каждого типа защит существуют тонкие градации устойчивости по отношению к условиям их удаления. Так, например, в группе сложных эфиров устойчивость к щелочному сольволизу возрастает в ряду: ChCCOO-R < C1CH2COO-R < CH3COO-R < C6H5COO-R < QHsNHCOO-R. Аналогично изменяется стабильность силиловых эфиров в условиях сольволиза в ряду: Me3Si-O-R < Me3CSi(Me2)—О—R < МезС81(Рп2)—О—R. Очень важной является возможность удаления силиль-ной группы при действии фторид-иона, что позволяет снимать эту группу, не затрагивая какие-либо другие защиты. В группе простых эфиров резко различными будут условия снятия защит при замене алкильной группы на ал-лильную, бензильную или тритильную. Так, удобным методом снятия ал-лильной защиты является двустадийная процедура: изомеризация в пропе-ниловый эфир под действием /я/>е/я-бутилага калия в абсолютном ДМСО (или под действием комплексов родия) и гидролиз в слабокислых условиях (см. схему 2.90). Бензильная группа может быть удалена либо в нейтральных условиях гидрогенолизом над палладиевым катализатором, либо путем од-ноэлектронною восстановления натрием в жидком аммиаке. Тритапьная и ее близкий аналог п-метокситритильная защиты очень сходны по своим свойствам, но они настолько сильно различаются по скорости кислотного сольволиза, что не представляет особой проблемы снятие и-метокситритиль-ной группы при сохранении тритильной.
Разнообразие методов защиты гидроксильной функции, равно как и способов удаления защитных групп, является мощнейшим инструментом, резко облегчающим решение всевозможных синтетических задач, так или иначе связанных с использованием спиртовых функциий. Среди них могут быть не только задачи, связанные с селективным получением тех или иных производных в ряду полигидроксильных соединений, как, например, показанная на схеме 2.89. В полном синтезе очень важным является применение системы защит, настроенной таким образом, чтобы сделать возможным использование полифункционального предшественника в качестве субстрата в последовательности контролируемых превращений, затрагивающих поочередно одну за другой эти функции.
Наглядным примером успешности такого подхода — подхода, стратегического по своему смыслу, — является синтез биологически активного природного дитерпеноида зоопатенола (212), выполненный Николау с сотр. [26е]. Ретросинтетический анализ этой структуры предполагал разборку по связям a, b и с, что позволило избрать в качестве основных синтетических блоков бромкетон 213 и триол 214 (схема 2.91). Формальный путь синтеза целевого продукта из этих исходных, включающий последовательность ряда превращений, также показан на схеме 2.91 (звездочками обозначены те центры в реактантах, которые участвуют в образовании связей на каждой из стадий).
С точки зрения общей стратегии этот план выглядит вполне убедительно, поскольку он включает сравнительно немного стадий, причем каждая из них предполагает использование хорошо известных реакций. Однако даже при поверхностном анализе становится ясным, что реализовать его в представленном виде просто невозможно из-за практически непреодолимых препятствий, обусловленных полифункциональным характером всех показанных реактантов 213—218 в этой гипотетической последовательности. Так, например, хотя чисто формально можно представить себе образование связи С-С при сборке 215 из предшественников 213 и 214 по схеме реакции Гриньяра между альдегидом, полученным окислением 214, и магнийорганическим соединением, приготовленным из бромида 213, но невозможно напрямую окислить 214 до альдегида требуемого строения, равно как и получить реагент Гриньяра из 213 (из-за присутствия карбонильного электрофила в этой молекуле). Легко убедиться, что столь же невозможной в действительности является реализация и других стадий показанной последовательности, несмотря на наличие хорошо отработанных методов для проведения этих превращений.
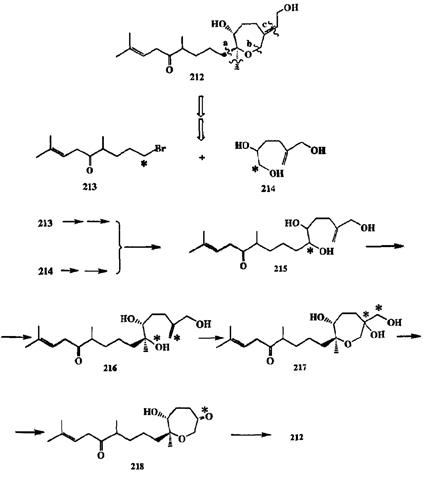 Схема 2.91
Схема 2.91
|
Очевидно, что было бы абсолютно бессмысленным делом попытаться реализовать хотя бы одну какую-либо из стадий этого плана с субстратами 213—218. Тем не менее, в действительности синтез 212 был успешно выполнен в полном соответствии с показанным выше планом и с использованием в качестве исходных веществ соединений 213 и 214, которые, однако, включались в синтетическую цепочку в виде защищенных производных (см. схему 2.92).
Синтетическим эквивалентом триола 214 послужило производное 219, в котором все три гицроксильные группы защищены по-разному. Селективное удаление тетрашдропиранильной защиты освобождает нужный первичный гидроксил, далее окисляемый до требуемого альдегида 220. Как уже отмечалось, кетобромид 213 не может непосредственно использоваться для получения соответствующего реагента Гриньяра. Однако ничто не мешает превратить 213 в соответствующий кеталь, из которого легко получить требуемый реагент 221. Реакция 220 с 221, последующее окисление единственной незащищенной гидроксильной группы продукта 222 и повторная реакция Гриньяра по получающейся карбонильной группе не представляют проблемы. Продукт 223 содержит две двойные связи, но лишь одна из них должна быть превращена в эпоксид, необходимый для последующего построения оксепанового цикла. Для эпоксидирования 223 нельзя использовать такие наиболее часто применяемые для этой цели реагенты, как надкислогы, ибо они в первую очередь будут атаковать более нуклеофильную трехзамещен-ную двойную связь. Для того чтобы обеспечить требуемую селективность окисления, была удалена силильная защита (действием несольватированного фтор-аниона), и полученный при этом аллиловый спирт окислен далее с помощью трет-ВuООН — реагента селективного эпоксидирования двойной связи в аллиловых спиртах. Ключевая стадия всего синтеза — внтримолеку-лярная циклизации эпоксида 224 с образованием семичленного цикла протекает вполне селективно, так как вторичный гидроксил, наиболее опасный конкурент реагирующей третичной гидроксильной группы, надежно защищен. Продукт циклизации диол 225 далее превращался в кетон 226 с помощью стандартного окисления 1,2-диольного фрагмента, после чего для завершения синтеза 212 необходимо было провести лишь несколько довольно тривиальных превращений.
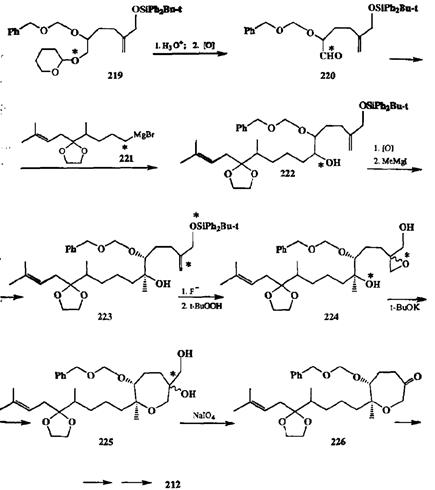 Схема 2.92
Схема 2.92
|
Очевидно, что успех всего синтеза определялся в первую очередь тщательно продуманным выбором системы защитных групп в исходных соединениях. Действительно, наличие трех разных защитных групп в 219, производном исходного триола 214, позволило удалять каждую из них именно в тот момент, когда требовалось провести то или иное превращение селективно с участием конкретной гидроксильной функции, а постановка защиты на кетонную функцию в бромиде 213 обеспечила сохранность кетонного фрагмента на всем протяжении синтетической последовательности. Примечательно, что в синтезе этой полифункциональной целевой структуры были сведены к минимуму манипуляции с защитными группами и не требовалось включения вспомогательных операций постановки и снятия дополнительных зашит на каких-либо стадиях.
До сих пор мы говорили о защищенных соединения как о производных, обеспечивающих сохранность той или иной функции в условиях синтетических превращений. Однако нередко одна и та же группировка может служить защитной в одной серии реакций и функциональной — в другой. Ниже будут рассмотрены некоторые примеры, иллюстрирующие важность этого аспекта использования защитных групп в синтезе.
Пожалуй, наиболее простым и очевидным является случай со сложноэфир-ной защитой спиртовой группы. Как мы уже отмечали выше, эта защита позволяет сохранить спиртовую функцию в условиях проведения таких реакций, как окисление или гликозилирование. Однако синтетически не менее важна способность сложных эфиров, особенно таких, кактрифторацетатыили трифлаты, служить активными электрофилами в реакциях с карбанионными нук-леофилами для образования связи С—С (см., например, схему 2.79).
Другой классический способ защиты спиртов — превращение их в трити-ловые эфиры. Наиболее часто этот способ используется для того, чтобы исключить возможность протекания электрофильного замещения водорода в соответствующей гидроксильной группы. Однако в случае вторичных спиртов переход к тритильным группы существенно облегчает отрыв гидрид-иона от а-СН-фрагмента под действием таких специфических катализаторов, как тритил-катион, в результате чего может достаточно легко происходить дис-пропорционирование с образованием кетонного фрагмента и трифенилме-тана. На схеме 2.93 приведен пример использования этой особенности три-тильной защиты для проведения селективного окисления вторичной спиртовой группы в бифункциональном субстрате 227 [26f].
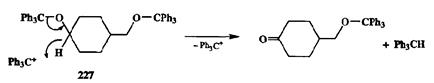 Схема 2.93
Схема 2.93
|
Хорошо известно, что превращение альдегидного карбонила в дитиоаце-тальную функцию обеспечивает сохранность этого карбонила в условиях реакций нуклеофильного присоединения, окисления или гидридного восстановления. Но не менее важным для синтеза является то обстоятельство, что дитиоацетали могут служить удобными предшественниками для генерации соответствующих карбанионных реагентов (под действием таких оснований, как бутиллитий), и в следующем разделе мы подробнее рассмотрим специфику этого применения дитиоацеталей.
Превращение кетонов в кетали — традиционный прием защиты этого фрагмента в условиях восстановления, особенно полезный в тех случаях, когда возможна селективная постановка этой защиты по одной из карбонильных групп субстрата. Так, монокеталь 228 (схема 2.94) легко и селективно можно получить из соответствующего дикетона, поскольку вторая кетонная группа (при С-17) в этом соединении стерически затруднена. Восстановление 228 боргидридом натрия дает (после гидролиза защитной группы) с почти коли-чественным выходом кетоспирт 229 — результат, можно сказать, банальный. Однако оказывается, что при восстановлении того же субстрата 228 может быть обеспечена с такойже полнотой и обратная региоселективность, а имен-но, исключительное восстановление по С-3 центру. Этот парадоксальный, на первый взгляд, результат достигается, если проводить восстановление с пoмощью дииодсилана, реагента специфического расщепления и гидрогеноли-за диоксолановой группировки [26g]. Таким образом, в реакции 228 → 230 ке-тальная группа (всего лишь замаскированный эквивалент кетогруппы!) вы-ступает в роли функции с довольно необычными свойствами.
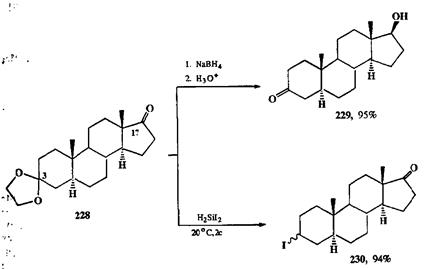 Схема 2.94
Схема 2.94
|
В ряду производных кислот особое место занимают амиды вследствие их пониженной электрофильности и, соответственно, повышенной стабильности в условиях методов, обычно применяемых для расщепления других карбоксильных производных. В целом, однако, амидная зашита используется не очень часто в синтезе именно в силу жесткости условий, требуемых для регенерации карбоксильной функции (см. примеры в работе [26g]). Тем не ме-нее, именно с использованием амидов удалось существенно упростить решение проблем селективности в реакции Михаэля в ряду производных а,р-не-предельных кислот. Так, известно, что взаимодействие эфиров таких кислот с магний- илилитийорганическими соединениями обычно приводит к образованию смесей продуктов 1,2- и 1,4-присоединения. В некоторых (но далеко не во всех!) случаях проблему селективного получения 1,4-аддуктов удается решить с помощью купратных реагентов. Ситуация резко упрощается, если брать диметиламиды типа 231(см. схему 2.95) в качестве акцепторов Михаэля. Благодаря наличию диметиламидного фрагмента полностью блокируется атака нуклеофила по карбонильному атому углероду, и реакции с ли-тийорганическими реагентами самой различной природы протекают исключительно как 1,4-присоединение [26h]. Более того, образующийся на первой стадии карбанионный интермедиат обладает достаточной стабильностью в условиях присоединения по Михаэлю, что дает возможность далее вводить его в реакции сшироким кругом элекгрофилов и таким образом получать набор разнообразных продуктов присоединения С-нуклеофилов и С-элекгрофилов по двойной связи субстрата типа 231.Того же результата можно добиться при работе с триметилгидразидами кислот, как, например, 232 [261].
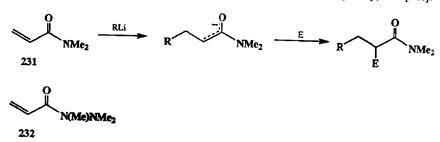 Схема 2.95
Схема 2.95
|
В настоящем разделе были изложены некоторые общие принципы применения защитных групп на примерах, относящихся к химии спиртовой и — в меньшей степени — карбонильной групп. К настоящему времени разработана весьма изощренная система защит почти всех главных функциональных групп [26c,d], и интенсивные исследования в этой области продолжаются. Так, в первом издании монографии по защитным группам (Green, «Protective Groups in Chemistry», 1981 г.) описано примерно 500 различных защит для пяти типов функциональных групп. К моменту публикации второго издания этой монографии в 1991 г. [26сJ к этому списку добавилось еще около 200 групп. Продолжающееся расширение этого набора более всего обусловлено постоянно возрастающим уровнем сложности синтетических задач, решение которых требует все более тонких инструментов для управления селективностью различных реакций в разнообразном структурном контексте.
Познакомившись с методами трансформаций функциональных групп и принципами обеспечения селективности реакций, мы можем теперь снова обратиться, но уже на более высоком уровне, к вопросам сборки углеродного скелета молекулы с учетом того, что в реальном синтезе все три задачи — создание связи С—С, обеспечение требуемой функциональности и эффективное управление селективностью реакций — решаются как единая комплексная проблема.
Дата добавления: 2015-04-05; просмотров: 2713;