Врожденные заболевания ВНЧС
Синдром Робена встречается на каждые 10—30 тыс. новорожденных. Этот синдром может быть частью более распространенной врожденной патологии
Врождённый порок ЧЛО, характерны три основные клинические признака:
 недоразвитие нижней челюсти (нижней микрогнатией),
недоразвитие нижней челюсти (нижней микрогнатией),
глоссоптоз (недоразвитие и западание языка)
наличие расщелины нёба.
У новорожденных и грудных детей этот синдром часто сопровождается нарушением двух важнейших функций:
1. Дыхания: на вдохе ребенок хрипит с втяжениями грудной клетки. Иногда ребенок не может дышать вообще и тогда в роддоме ребенку в трахею устанавливают интубационную трубку для обеспечения дыхания. Общее название такого нарушение дыхания – синдром обструктивного апноэ, который является жизнеугрожающим состоянием и может быть причиной синдрома внезапной смерти.
2. Глотания. При этом питание может осуществляться только через желудочный зонд.
С такими нарушениями новорожденные дети прямо из родильного отделения переводятся в отделение реанимации, где проводится длительное выхаживание ребенка. Для предупреждения сердечной недостаточности вследствие хронической обструкции дыхательных путей используются положение ребенка ничком, прошивание языка или трахеостомия. Для преодоления проблем с питанием используется наложение гастростомы под местной анестезией. Интубация может быть крайне затруднена, а иногда и невозможна даже при наличии волоконно-оптических инструментов и другой техники. У больных с этим синдромом анестезия проводится только опытными анестезиологами
Между тем, длительное пребывание ребенка на трубке и зондовом питании чревато развитием пневмонии, ослаблением глотательного рефлекса. А срок выхаживания может быть неопределенно долгим, пока ребенок не вырастет и просвет верхних дыхательных путей позволит ему самостоятельно дышать и питаться (обычно к 5-6 месяцам).
При синдроме Пьера Робена всему виной недоразвитие (укорочение) нижней челюсти, из-за чего язык смещен назад и перекрывает просвет дыхательных путей (Рис. 1). Если удлинить нижнюю челюсть, то язык переместится за ней вперед и откроет дыхательные пути (Рис. 2).
Существует метод, позволяющий удлинить нижнюю челюсть – компрессионно-дистракционный остеосинтез.
Метод заключается в том, что на нижнюю челюсть с 2-х сторон устанавливаются аппараты, проводятся распилы челюсти между лапками аппаратов и фрагменты челюсти плотно прижимаются друг к другу — компрессия (Рис. 3). С 5-6 суток после операции фрагменты челюсти постепенно, со скоростью 1 миллиметр в сутки, разводятся аппаратами на необходимую величину. Процесс удлинения челюсти аппаратами называется дистракцией, а аппарат соответственно – компрессионно-дистракционным. Между фрагментами кости образуется новая молодая кость – костная мозоль (Рис. 4). Операция проводится под интубационным наркозом. Такое оперативное лечение у новорожденных и младенцев проводится по жизненным показаниям.
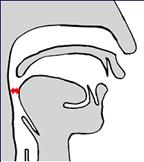 Рис. 1. При недоразвитии н/ч язык смещен кзади и перекрывает просвет верхних дыхательных путей
Рис. 1. При недоразвитии н/ч язык смещен кзади и перекрывает просвет верхних дыхательных путей
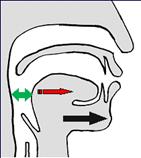 Рис. 2. В результате дистракции н/ч перемещается кпереди вместе с языком, который перестает перекрывать верхние дыхательные пути.
Рис. 2. В результате дистракции н/ч перемещается кпереди вместе с языком, который перестает перекрывать верхние дыхательные пути.
Самостоятельное дыхание становится возможным в среднем уже на 4-6 сутки дистракции, которая продолжается до достижения правильного соотношения челюстей. На самостоятельное питание ребенок переводится, как правило, к моменту окончания дистракции или немного позже. Ребенок выписывается с аппаратами на время (3 месяца) превращения костной мозоли в полноценную кость. Через 3 месяца – повторная госпитализация для удаления аппаратов, которое также проводится под наркозом.
Поскольку страховой полис и финансирование больницы не покрывают стоимость аппаратов, их необходимо приобретать отдельно. Их стоимость – 250-300 € за аппарат отечественного предприятия КОНМЕТ и 1200-1500 € за аппарат немецкого предприятия MARTIN. Все остальное лечение (в том числе и операция) проводятся бесплатно.
Важно отметить, что синдром обструктивного апноэ может наблюдаться и у детей более старших возрастов при заболеваниях, которые сопровождаются недоразвитием костей лицевого скелета: синдромы Франческетти (син.: с. Тричера-Коллинза), I-II жаберных дуг (син.: гемифациальная микросомия, с. Гольденхара), Апера, Крузона и др.; артрозах и анкилозах височно-нижнечелюстных суставов, сопровождающихся недоразвитием нижней челюсти.
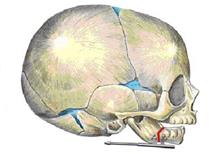
Рис. 3. Способ установки аппарата. Красным показана плоскость распила.
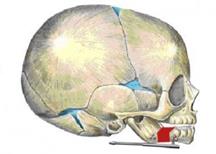 Рис. 4. Результат удлинения н/ч. Красный участок – костная мозоль между фрагментами нижней челюсти.
Рис. 4. Результат удлинения н/ч. Красный участок – костная мозоль между фрагментами нижней челюсти.
КЛИНИЧЕСКИЙ ПРИМЕР. В отделение реанимации из родильного дома поступил ребенок 2-х недель жизни с диагнозом синдром Пьера Робена, синдром обструктивного апноэ тяжелой степени. Питание может осуществляться только через зонд. У ребенка имеется дефицит веса. В связи с выраженными нарушениями дыхания и питания ребенку проведен компрессионно-дистракционный остеосинтез нижней челюсти.
После окончания дистракции ребенок выписан на самостоятельном дыхании и питании.
Синдром Гольденхара или I-II жаберных дуг (син.: гемифациальная микросомия)
Триада врожденных аномалий, состоящая из:
· эпибульбарных дермоидов (двусторонних, у лимба или роговичного края глаза),
· множественных преарикулярных аппендиксов,
· претрагусных свищей.
Кроме того, часты сочетания с:
· аномалиями позвоночника,
· односторонней гипоплазией нижней челюсти,
· односторонней микроотией, атрезией или стенозом наружного слухового прохода, обычна односторонняя тугоухость проводящего типа.
· колобома глаз. Однако встречаются и дефекты внутреннего глаза.
Goldenhar синдром правой ушной дисплазии ушной раковины, при наличии предварительного ушной apêndices
Синдром БЕРРИ-ФРАНЧЕСКЕТТИКОЛЛИНЗА (син.: синдром Франческетти, челюстно-лицевой дизостоз). Врожденная патология, чаще встречаются спорадические случаи.
Наследуется по аутосомно-доминантному типу, чаще встре чаются спорадические случаи.
Характеризуется гипоплазией нижней челюсти,
монголоидным типом лица,
наличием колобомы (врожденный или приобретенный дефект глаза, приводящий к различным аномалиям: от возникновения небольшого углубления края века или нижней части радужной оболочки),
деформацией ушных раковин,
глухотой,
макрогнатией.
Нередко сочетается с деформацией конечностей, отсутствием лучевых костей,
радиоульнарным синостозом (врожденном сращении проксимальных участков локтевой и лучевой кости) и гипоплазией или отсутствием I пальцев кистей.
Проводят дифференциальный диагноз с синдромом Апера и синдромом Крузона.
Лечение: при возможности - пластическая операция.
Вопросы для контроля исходного уровня знаний студентов
1. Строение ВНЧС в возрастном аспекте.
2. Классификация заболеваний сустава в детском и подростковому возрасте по А.А. Колесову и Ю.І.Бернадскому.
3. Врожденные заболевания ВНЧС.
4. Причины возникновения артритов и остеоартритив ВНЧС у детей.
5. Острый и хронический артрит. Клиника, диагностика, дифференциальная диагностика.
6. Лечение острых и хронических артритов ВНЧС. Профилактика осложнений, результат заболевания.
7. Клиническая картина при юношеских заболеваниях ВНЧС.
8. Клинические и ретнгенологические признаки деформирующего остеоартроза.
9. Клинический и рентгенологический признаки анкилоза ВНЧС.
10. Принципы лечения деформирующего остеоартроза и анкилоза ВНЧС.
Дата добавления: 2015-02-10; просмотров: 6760;