Внутреннее вращение и поворотная изомерия
Классическая органическая химия предполагала, что вращение атомных групп вокруг единичных связей происходит совершенно свободно. Тем самым любые конформации, например этана, возникающие в результате внутренних поворотов, имеют одинаковую свободную энергию (изменение угла поворота не требует затраты энергии) На рис.1 изображено вращение вокруг связи С-С в молекуле этана (CH3-CH3):

|
| Рис.1 Расположение С-С связей этана в транс-(a), цис-(б) и гош-(свёрнутой) (в) конформациях. (Проекция на плоскость, перпендикулярную с-с связям). |
Однако исследования термодинамических свойств этана и других соединений с единичными связями, а также структурные исследования, проведенные методами спектроскопии, показали, что внутреннее вращение всегда несвободно. Молекула этана имеет минимум энергии в скрещенной, или транс-конформации, и максимум – в цис-конформации.
Для поворота на 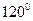 ,т.е. для перехода из одной транс- конформации в другую, ей тождественную, нужно преодолеть барьер (энергетический) равный 2,9 ккал/моль.
,т.е. для перехода из одной транс- конформации в другую, ей тождественную, нужно преодолеть барьер (энергетический) равный 2,9 ккал/моль.
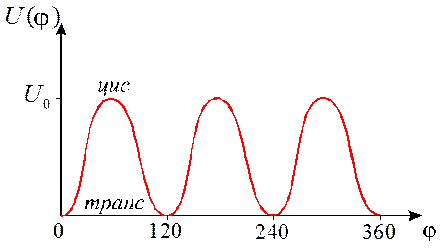
|
| Рис.2 График зависимости потенциальной энергии внутреннего вращения в этапе от угла поворота. (U0=2, 9 ккал/моль) |
Для этана и для других молекул с осевой симметрией С3 зависимость потенциальной энергии внутреннего вращения U от угла поворота 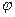 можно приближенно представить формулой:
можно приближенно представить формулой: 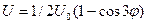 , где
, где 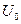 - высота потенциального барьера. Значение возрастает при замене атомов Н на более объемистые атомы и группы, например, для CH3-C(CH3)3 – U0=4,4 ккал/моль. уменьшается при удлинении оси вращения.
- высота потенциального барьера. Значение возрастает при замене атомов Н на более объемистые атомы и группы, например, для CH3-C(CH3)3 – U0=4,4 ккал/моль. уменьшается при удлинении оси вращения.
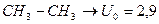 ккал/моль.
ккал/моль. 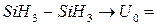 1 ккал/моль.
1 ккал/моль.
Потенциальная энергия внутреннего вращения определяется взаимодействием валентно не связанных атомов и групп. Строгий квантово-механический расчет U затруднен, т.к. она определяется как малая разность полных энергий молекулы в цис- и транс- конформациях. Тормозящий потенциал (или барьер) возникает вследствие стерического, ван-дер-ваальсового отталкивания валентно не связанных атомов и квантово- механического взаимодействия связей, примыкающих к оси вращения (эффект ориентации связей). И то, и другое делает более устойчивой транс- конформацию (принцип скрещенных связей).
Отталкивание можно оценить, зная характерные кривые зависимости энергии межмолекулярного взаимодействия от межмолекулярного расстояния для модельных веществ.
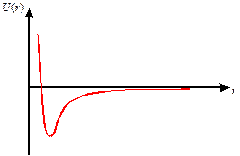
| Рис.3 Зависимость энергии межмолекулярного взаимодействия от расстояния. |
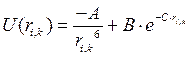 - потенциал Букингема.
- потенциал Букингема.
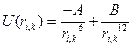 - потенциал Леннарда-Джонса.
- потенциал Леннарда-Джонса.
Эффект ориентации связей определить трудно. Грубую оценку можно получить, считая, что в этане из-за малого ван-дер-ваальсового радиуса атома Н 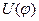 целиком определяется эффектом ориентации, и что у производных этана и у него самого этот эффект одинаков. Тогда для производных этана получают
целиком определяется эффектом ориентации, и что у производных этана и у него самого этот эффект одинаков. Тогда для производных этана получают 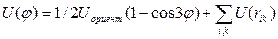 ,
,
где 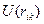 - "стерическая" потенциальная энергия взаимодействия валентно не связанных атомов i и k, находящихся на расстоянии
- "стерическая" потенциальная энергия взаимодействия валентно не связанных атомов i и k, находящихся на расстоянии 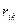 друг от друга. зависит от . Если молекула содержит сильно полярные связи, то к правой части последнего выражения надо добавить члены, учитывающие электростатическое взаимодействие атомов связей, примыкающих к оси вращения. Это существенно для биополимеров. Потенциал взаимодействия атомов Н и С связей С-Н и С-С описывается эмпирическими потенциалами Хилла, Бартелла, Китайгородского и др. на основе данных о кристаллохимических и термодинамических свойствах простых углеводородов. Эти потенциалы имеют свой вид
друг от друга. зависит от . Если молекула содержит сильно полярные связи, то к правой части последнего выражения надо добавить члены, учитывающие электростатическое взаимодействие атомов связей, примыкающих к оси вращения. Это существенно для биополимеров. Потенциал взаимодействия атомов Н и С связей С-Н и С-С описывается эмпирическими потенциалами Хилла, Бартелла, Китайгородского и др. на основе данных о кристаллохимических и термодинамических свойствах простых углеводородов. Эти потенциалы имеют свой вид 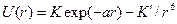 , где а, K, K`- const.
, где а, K, K`- const.
Китайгородский ввел универсальную функцию 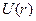 так называемый атом-атомный потенциал. В случае пары атомов он выражает взаимодействие "универсальных нейтральных атомов" и характеристических зарядов на ядрах. Функция Китайгородского имеет вид:
так называемый атом-атомный потенциал. В случае пары атомов он выражает взаимодействие "универсальных нейтральных атомов" и характеристических зарядов на ядрах. Функция Китайгородского имеет вид:
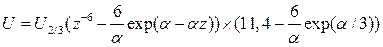 ,
,
где 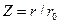 ,
, 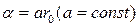
r0 – сумма Ван-дер-Ваальсовых радиусов взаимодействия атомов, 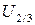 - значение U при r=2/3r0.
- значение U при r=2/3r0.
Для связей типа С-С, С-H, H-H Китайгородский принял значение =3.5ккал/моль, α=13. При этих значениях 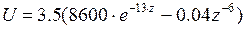 . Приведенное выражение дает хорошие результаты при вычислении конформаций как малых, так и макромолекул.
. Приведенное выражение дает хорошие результаты при вычислении конформаций как малых, так и макромолекул.
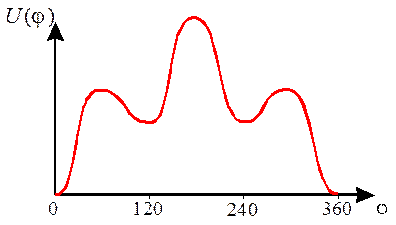
Если молекула лишена аксиальной симметрии, то кривая U(φ) становится несимметричной и приведенное выражение для U уже непригодно. Так для бутана ( 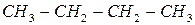 ) U(φ) имеет вид:
) U(φ) имеет вид:
|
| Рис.4 Зависимость энергии от угла вращения для бутана |
Очевидно, что молекулы, характеризуемые несколькими неэквивалентными минимумами энергии U(φ),будут существовать именно в этих состояниях, переходя из одной конформации в другую со скоростями, определяемыми высотами барьеров, разделяющих минимумы.
Относительное равновесное содержание молекул Н-бутана в гош- и транс- конформациях выражается величинами:
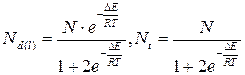
где Nt-число молекул в транс конформации, Nd и Nl-число молекул в конформациях, свёрнутых вправо и влево на 120˚
Очевидно, что Nt+Nd+Nl=N (N-полное число молекул). Таким образом, вещество представляет собой динамическую смесь конформаций, которую в этих случаях, принято называть поворотными изомерами или ротамерами, или конформерами.
Состав термодинамически равновесной смеси определяется разностью внутренних энергий поворотных изомеров и температурой. При 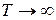 Nt=Nd =Nl= 1/3 N. При понижении температуры вещество кристаллизуется в форме одного наиболее устойчивого ротамера.
Nt=Nd =Nl= 1/3 N. При понижении температуры вещество кристаллизуется в форме одного наиболее устойчивого ротамера.
При высотах барьера порядка нескольких ккал/моль время поворотной изомеризации, т.е. время превращения одного ротамера в другой ~10-10сек. (получено на основе теории абсолютных скоростей реакции). За такие времена ротамеры не могут быть отделены друг от друга. Наличие ротамеров в равновесной смеси устанавливается путем изучения химических и физических свойств. Пространственное строение ротамеров различно, значит, различаются и их спектры. Спектр вещества представляет собой результат наложения спектров ротамеров. За время жизни ротамера происходят приблизительно сотни и тысячи колебаний (частоты ~1012-1013 сек-1,при времени поворотной изомеризации ~10-10сек).
Впервые существование поворотной изомерии было установлено Кольраушем с помощью спектров комбинационного рассеяния. Отношение интенсивности спектральных линий, отвечающих различным ротамерам, зависит от их содержания в смеси. Оно меняется с температурой и, следовательно, разности энергии ротамеров E можно определить по температурному ходу интенсивностей спектральных линий. Так для Н-бутана E~0,6 ккал/моль. Информацию по ротамерии получают также с помощью ЯМР, электронографии, измерения дипольных моментов молекул и т.д.
Теоретический расчет величин Е можно провести с помощью потенциалов Китайгородского, Хилла и др. на полуэмпирической основе. Для молекул типа Н-бутана и более сложных приходится учитывать повороты вокруг нескольких связей. Энергия внутреннего вращения зависит соответственно от нескольких углов вращения и изображается уже не кривой, а поверхностью (вообще говоря, многомерной).
Изучение поворотной изомерии конформационных превращений молекул приобрело сейчас очень большое значение в органической и биоорганической химии. Химические и физико-химические свойства молекул существенным образом зависят от их конформаций. Главные особенности физического поведения макромолекул определяются поворотной изомерией.
Дата добавления: 2015-02-07; просмотров: 3992;