Нарушения фенотипического пола
Женский псевдогермафродитизм
Врожденная гиперплазия надпочечников. Клинические проявления. Пути образования глюкокортикоидов в надпочечниках и андрогенов в яичках и надпочечниках показаны на рис. 333-3. Три фермента принимают участие в синтезе как глюкокортикоидов, так и андрогенов (20,22-десмолаза, 3-гидроксистероид-дегидрогеназан 17-гидроксилаза); недостаточность любого из них препятствует образованию глюкокортикоидов и андрогенов и, следовательно, приводит как к врожденной гиперплазии надпочечников (из-за повышения уровня АКТГ), так и к недостаточной вирилизации эмбриона мужского пола (мужской псевдогермафродитизм). В синтезе андрогенов участвуют два фермента— 17,20-десмолаза и 17-гидроксистероид-дегидрогеназа; недостаток какого-либо из них приводит к чистому мужскому гермафродитизму при нормальном синтезе глюкокортикоидов. Дефицит любого из двух последних ферментов синтеза глюкокортикоидов (21-гидроксилаза и 11-гидроксилаза) нарушает образование гидрокортизона; компенсаторно возрастающая секреция АКТГ вызывает гиперплазию надпочечников и вторичное усиление выработки андрогенов, что приводит к вирилизации у женщин и преждевременной маскулинизации у мужчин.
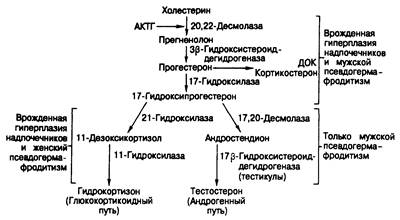
Рис. 333-3. Пути синтеза глюкокортикоидов и андрогенов.
Недостаточность надпочечников при этих нарушениях обусловливает тяжелую и угрожающую жизни патологию у лиц обоего пола. Подробно это рассматривается в гл. 325. Основные особенности разных форм врожденной гиперплазии надпочечников перечислены в табл. 333-4. Рассматривая нарушения полового развития, целесообразно проанализировать ферментные нарушения в стероидогенезе, приводящие либо к женскому, либо к мужскому псевдогермафродитизму. (Одно из таких нарушений — недостаточность 3-гидроксистероид-дегидрогеназы — обусловливает как мужской, так и женский гермафродитизм, но, поскольку более распространенным пороком развития половых органов является неполная вирилизация у мужчин, эта ферментная патология рассматривается здесь как нарушение дифференцировки мужского фенотипа.)
Чаще всего причиной амбисексуальности половых органов у новорожденных служит врожденная гиперплазия надпочечников вследствие недостаточности 21-гидроксилазы (в Европе она встречается с частотой 1:5000, а в США — 1:15 000). Вирилизация у девочек проявляется обычно уже при рождении, а у мальчиков — в первые 2—3 года жизни. Для девочек характерны гипертрофия клитора в сочетании с его вентральным подтягиванием (патологическая эрекция), частичное сращение лабиоскотальных складок и вирилизация уретры различной степени. Внутренние женские половые органы и яичники остаются интактными, а вольфовы протоки регрессируют нормально, вероятно, потому, что надпочечники начинают функционировать на относительно поздних этапах эмбриогенеза. Наружные половые органы у девочек сходны с таковыми у мальчиков с двусторонним крипторхизмом и гипоспадией. Лабиоскротальные складки увеличены и морщинисты и напоминают мошонку. В редких случаях вирилизация достигает такой степени, что у девочки полностью развивается мужская уретра, половой член, а также предстательная железа, что приводит к ошибке при определении пола новорожденных. При рентгенографии после введения контрастного вещества в наружное половое отверстие обнаруживают влагалище, матку и иногда даже маточные трубы. В немногих случаях вирилизация девочек при рождении выражена незначительно или вообще отсутствует и проявляется лишь позднее — в детстве, отрочестве или зрелости. По-видимому, это связано с аллельной вариацией мутантных генов (так называемая поздно проявляющаяся, или взрослая, форма нарушения). Без лечения больные девочки в течение первого года жизни быстро растут и вирилизация у них прогрессирует. В период ожидаемого пубертата нормального полового созревания по женскому типу не происходит и менструации не появляются. Быстрое соматическое созревание у лиц обоего пола приводитк преждевременному заращению эпифизарных щелей и низкорослости в зрелые годы.
Таблица 333-4. Формы врожденной гиперплазии надпочечников
| Дефект | Кортизол | Альдостерон | Степень вирилизации у женщин | Степень недостаточной вирилизации у мужчин | Преимущественно секретируемый стероид | Примечания |
| Частичная недостаточность 21-гидр оксидазы (простая вирилизирующая или компенсирования форма) | Норма | | ++++ | 17-Гидроксипрогестерон | Самая распространенная форма (примерно 95% всех случаев); у 1/3—2/3 больных отмечается потеря соли | |
| Тяжелая недостаточность 21-гидроксплазы (сопр ов ождается потерей соли) | | | ++++ | 17-Гидроксипрогестерон | ||
| Недостаточность 1 lp-гид-роксилазы (гипертоническая форма) | | | ++++ | 11-Дезоксп-кортизоли 11-дезоксикорти-костерон | Гипертензия | |
| Недостаточность Зр-гпд-рокснстеропд-дегидрогеназы | о | + | ++++ | Д^Зр-ОН-сое-динения (дегидроэпиандростерон) | По-видимому, вторая по частоте встречаемости; обычно протекает с потерей соли | |
| Недостаточность 17-гндроксидазы | | | ++++ | Кортикостерон и 11-дезоксикортикостерон | Отсутствие феминизации у женщин, гипертензия | |
| Недостаточность 20,22-десмолазы (липоидная гиперплазия надпочечников) | о | о | ++++ | Холестерин (?) | Редко встречающаяся форма; обычно протекает с потерей соли |
Так как дифференцировка мужского фенотипа остается нормальной, заболевание у мальчиков при рождении обычно не распознается, если только нет явной недостаточности надпочечников. Однако уже в первые годы жизни у больных наблюдают интенсивный рост и созревание наружных половых органов, частые эрекции и чрезмерное развитие мышц. Вирилизация у мальчиков может проявляться двояко. Избыточная секреция андрогенов надпочечниками ингибирует продукцию гонадотропинов, так что яички остаются незрелыми, несмотря на ускорение маскулинизации. В зрелые годы такие больные, если их не лечить, способны к эрекции и эякуляции, но сперматогенез у них отсутствует. В других случаях секреция андрогенов надпочечниками может активировать преждевременное созревание гипоталамо-гипо4шзарной оси и инициировать истинное преждевременное половое созревание, включая и сперматогенез (см. гл. 330). У нелеченых мужчин могут появляться АКТГ-зависимые «опухоли» яичек, состоящие из остатков клеток надпочечников.
При недостаточности 21-гидроксилазы, обусловливающей около 95% случаев врожденной гиперплазии надпочечников, продукция гидрокортизона уменьшается и, следовательно, возрастает секреция АКТГ, усиливается рост надпочечных желез, и тем самым происходит частичная или полная компенсация нарушения секреции гидрокортизона. Примерно у 50% больных отмечают частичную недостаточность фермента, и секреция кортизола остается нормальной. Эта форма заболевания называется простой вирилизирующей, или компенсированной. У остальных имеет место более полный дефицит фермента; даже увеличенные надпочечники не могут продуцировать адекватное количество кортизола и альдостерона, что приводит к выраженной потере соли с анорексией, рвотой, уменьшением объема жидкости и коллапсу в первые недели жизни. Это чак называемая форма недостаточности 21-гидроксилазы с потерей соли. У всех нелеченых больных отмечают избыточную продукцию предшественников кортизола, образующихся до стадии, катализируемой 21-гидроксплазой, в силу чего в плазме возрастает содержание прогестерона и 17-гидроксипрогестерона. Они действуют как слабые антагонисты альдостерона на рецепторном уровне, что при компенсированной форме требует большей, чем в норме, продукции альдостерона, чтобы сохранить нормальный баланс натрия.
Женский псевдогермафродитизм может быть вызван и недостатком 11-гидроксилазы. В этом случае блокада гидроксилирования 11-го углеродного атома приводит к накоплению 11-дезоксикортизола и дезоксикортикостерона (ДОК) — сильного сользадерживающего гормона, что сопровождается не потерей соли, а гипертензией. Клинические проявления, обусловливаемые дефицитом глюкокортикоидов и избытком андрогенов, сходны с таковыми при недостаточности 21-гидроксилазы.
Патофизиология. Оба нарушения — следствие аутосомно-рецессивной мутации. Частота недостаточности 21 -гидроксилазы составляет примерно 1:50. Идентифицировано не менее трех форм недостаточности этого фермента, и все они связаны с мутациями генов, расположенных на 6-й хромосоме вблизи от локуса HLA-B: наиболее распространенный тип, проявляющийся как обычная аутосомно-рецессивная мутация с изменением ферментной активности: вариант, обусловленный криптическим аллелем, который даже у гомозигот не имеет никаких клинических проявлений, но вызывает типичное заболевание, если сосуществует с распространенным вариантом, и поздно проявляющийся вариант. Носители заболевания (равно как и гомозиготы) среди членов данной семьи могут быть идентифицированы но гаплотипу HLA. При недостаточности 11-гндрокснлазы связь мутации с системой HLA остается неизвестной.
Эндокринная патология при этом состоянии рассматривается в гл. 325. Вкратце она сводится к повышению экскреции кетостероидов, как и основных метаболитов, накапливающихся выше места ферметной блокады У нелеченых больных повышено содержание АКТГ в плазме. При недостаточности 21-гидроксилазы в крови накапливается 17-гидроксипрогестерон, выводимый с мочой преимущественно в виде прегнантриола. При недостаточности 11-гидроксилазы в крови накапливается 11-дезокснкортизол, который выводится с мочой преимущественно в виде тетрагидрокортсксолона.
Лечение. Выбор пола должен определяться хромосомным и гонадиым полом, и соответствующую хирургическую коррекцию наружных половых органов следует производить как можно в более ранние сроки. Это весьма важно, так как при правильном лечении мужчины и женщины могут стать фертильными. Однако, если правильный диагноз устанавливается поздно (в возрасте старше 3 лет), выбор пола следует производить лишь после тщательного учета психосексуалыюй ориентации.
Консервативное лечение глюкокортикоидами предотвращает проявления недостаточности гидрокортизона, останавливает быструю вирилизацию и препятствует преждевременному соматическому развитию и заращению эпифизов. При недостаточности 11-гидроксилазы подавление патологической секреции стероидов приводит к нормализации артериального давления, а при обоих вариантах обеспечивает своевременное начало менструальной функции и развитие женских вторичных половых признаков. У мужчин терапия глюкокортикоидами подавляет секрецию андрогенов надпочечниками и приводит к нормализации секреции гонадотропинов, развитию яичек и сперматогенезу. Контролируют заместительную терапию, определяя содержание в плазме 17-гидропрогсстерона, андростендиона. АКТГ и ренина. При тяжелых формах 21-гидроксилазной недостаточности, сопровождающейся потерей соли или повышением активности ренина в плазме, показано и лечение минералокортикоидами. У таких больных об адекватности заместительной минералокортикоидной терапии судят по активности ренина в плазме. Женский псевдогермафродитизм вненадпочечниковою генеза.Женский псевдогермафродитизм редко имеет вненадпочечниковые причины. В прошлом введение беременным женщинам для профилактики аборта прогестинов, обладающих побочными андрогенными эффектами (таких как 17-этннил-19-нортестостерон), приводило к маскулинизации плодов женского пола. Женский псевдогермафродитизм может встречаться также у детей, рожденных матерями с вирилизирующими опухолями (например, арренобластомами или лютеомами беременных); в редких случаях причину заболевания установить не удается.
Врожденные дефекты мюллеровых протоков (врожденное отсутствие влагалища, агенезия мюллеровых структур). Клинические проявления. Врожденная гипоплазия, или отсутствие влагалища, в сочетании с аномалией или отсутствием матки (синдром Майера — Рокитанского — Кюстера — Хаузера) в качестве причины первичной аменореи уступает только дисгенезии гонад. У большинства больных нарушение диагностируют в возрасте ожидаемо! о полового созревания в связи с отсутствием менструаций. Рост и психическое развитие у них нормальны, а молочные железы, подмышечное и лобковое оволосение, а также телосложение соответствуют женскому типу. Матка может быть почти нормальной, лишенной только наружного входного канала, но чаще представлена рудиментарными двурогими тяжами с просветом или без него. У некоторых больных в области живота периодически появляются боли, что указывает на наличие достаточно функционального эндометрия, чтобы вызвать ретроградную менструацию и/или гематометру.
Примерно у 30% больных выявляют аномалии почек, чаще всего агенезию или эктопию. Встречается также сращение почек в виде подковы и солитарные эктопические почки, расположенные в тазовой полости. У 10% больных имеются нарушения скелета, причем у 60% из них в процесс вовлекается позвоночник, а у остальных — конечности и ребра. Специфические костные изменения характеризуются заклиниванием позвонков, их слиянием, рудиментарностью или асимметричностью тел позвонков и наличием дополнительных позвонков. Часто при этом наблюдают синдром Клнппеля—Фейля (врожденное сращение шейных позвонков, короткая шея, низкая задняя линия оволосения, а также болезненность и ограниченность движений шейного отдела позвоночника).
Патофизиология. Все больные имеют кариотип 46, XX. Чаще всего болезнь возникает спорадически, хотя наблюдали и несколько семейных случаев. Характер наследования в большинстве семейных случаев соответствует ограниченной полом аутосомно-доминантной мутации. Неясно, представляют ли собой спорадические случаи новые мутации того же типа, который определяет семейное нарушение, или они имеют многофакторную причину. Для семейных случаев характерна непостоянная экспрессируемость; у некоторых пораженных членов семьи имеются лишь скелетные или почечные аномалии, тогда как у других наблюдаются иные нарушения в производных мюллеровых протоков, например удвоение матки.
У мертворожденных плодов отсутствие матки и влагалища часто сочетается с двусторонней аплазией почек. Поэтому во всех случаях следует интересоваться наличием в семейном анамнезе изолированных нарушений скелета или ночек, а также мертворождений, которые могли бы быть связаны с врожденным отсутствием у плода обеих почек.
О сохранности функции яичников свидетельствуют овуляторные пики уровня ЛГ в плазме и двухфазные температурные кривые в течение цикла. У больных с нормальной маткой после хирургической пластики влагалища возможна беременность.
Лечение. Больных с агенезией влагалища можно лечить хирургически и консервативно. Цель хирургического вмешательства — создать искусственное влагалище путем имплантации резинового канала, покрытого несколькими слоями кожи. Консервативное лечение заключается в повторном давлении простым расширителем на влагалищную ямку, чтобы обеспечить ее достаточную глубину. Поскольку общая частота осложнений при хирургическом лечении составляет 5— 10%, у большинства больных следует пытаться использовать консервативный подход. Хирургическое же вмешательство можно рекомендовать женщинам с хорошо сформированной маткой, когда сохраняется возможность беременности. Чтобы сохранить новообразованное любым методом влагалище больной, целесообразно вести регулярную половую жизнь или проводить инструментальное расширение органа.
Дата добавления: 2015-03-17; просмотров: 918;