Схема 31. Патогенез системной красной волчанки
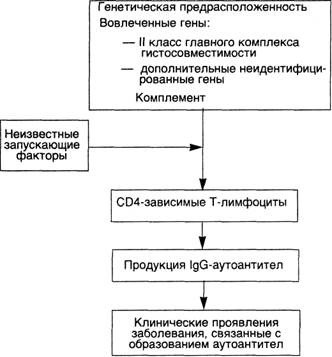
Генетические факторы.Члены семьи больного СКВ имеют повышенный риск развития заболевания. Примерно у 20 % ближайших родственников, не имеющих клинических проявлений СКВ, находят аутоантитела и другие нарушения в иммунной регуляции. Обнаружена более высокая конкордантность (24 %) у монозиготных близнецов по сравнению с дизиготными (1—3 %), причем у монозиготных близнецов, дискордантных по СКВ, виды и титры аутоантител похожи. Видимо, существует генетическая регуляция образования аутоантител, но развитие болезни (тканевые повреждения) зависит от негенетических факторов. Известно, что именно гены главного комплекса гистосовместимости (ГКГС) регулируют продукцию специфических аутоантител. Некоторые больные СКВ имеют врожденный дефицит компонентов комплемента, таких как С2 или С4. Отсутствие компонентов комплемента нарушает элиминацию циркулирующих иммунных комплексов системой мононуклеарных фагоцитов и способствует их осаждению в тканях.
Негенетические факторы.Доказано, что некоторые лекарства (гидролазин, прокаинамид, D-пеницилламин) могут вызывать СКВ-подобный ответ у человека. Ультрафиолетовое облучение обостряет течение заболевания у многих больных, видимо, благодаря способности УФ-лучей влиять на иммунный ответ. Под действием УФ-лучей кератиноциты продуцируют ИЛ-1.
Иммунологические факторы.Полагают, что в основе СКВ лежит гиперактивность В-лимфоцитов. Установлено также, что Т-лимфоциты-хелперы, выделенные из периферической крови больных СКВ, способны индуцировать in vitro секрецию анти-ДНК-антител аутологичными В-лимфоцитами. Эти анти-ДНК-антитела являются катионами и способны осаждаться в почечных клубочках.
Большинство висцеральных повреждений при СКВ обусловлено иммунными комплексами (III тип реакций гиперчувствительности). ДНК-антиДНК-комплексы определяются в клубочках почек и мелких кровеносных сосудах. При появлении аутоантител против эритроцитов, лейкоцитов и тромбоцитов развивайся реакция гиперчувствительности II типа.
Таким образом, системная красная волчанка представляет собой сложное мультифакториальное заболевание, развивающееся
в результате взаимодействия генетических, гуморальных факторов и факторов окружающей среды, которые, действуя совместно, вызывают активацию хелперных Т- и В-лимфоцитов, что способствует секреции различных видов аутоантител.
Морфологические изменения.Исключительно вариабельны. Патогномоничные морфологические изменения практически отсутствуют. При постановке диагноза необходимо учитывать клинические, серологические и морфологические данные. Наиболее характерным повреждением считается выпадение иммунных комплексов, которые находят в кровеносных сосудах, почках, соединительной ткани и коже.
Синдром Шегрена.Характеризуется сухостью глаз (сухой кератоконъюнктивит) и рта (ксеростомия), возникающими в связи с иммунологически обусловленной деструкцией слезных и слюнных желез. Он протекает как изолированное заболевание (первичная форма, или болезнь Шегрена), однако чаще связан с другими аутоиммунными заболеваниями (вторичная форма). Среди этих заболеваний чаще всего встречаются ревматоидный артрит, а также СКВ, полимиозит, склеродермия, васкулит, смешанные заболевания соединительной ткани и тиреоидит.
Этиология и патогенез.Морфологически наблюдаются лимфоцитарная инфильтрация и фиброз серозных и слюнных желез. В инфильтрате содержатся преимущественно активированные СО4+Т-лимфоциты-хелперы, а также В-лимфоциты, включая плазматические клетки, которые местно секретируют антитела. Остается до конца неясным, опосредованы ли тканевые повреждения только цитотоксическими Т-лимфоцитами, инфильтрирующими железы, или аутоантителами, небольшое количество которых находят в сыворотке крови.
Как и при других аутоиммунных заболеваниях, при синдроме Шегрена имеется ассоциация со II классом аллелей HLA.
В целом развитие синдрома Шегрена связывают с наличием нескольких типов аутоантител, хотя их спектр и не так широк, как при СКВ. Наиболее важными серологическими маркерами этого заболевания являются антитела против двух РНП-антигенов SS-A (Ro) и SS-B (La), которые выявляются у 90 % больных.
Прогрессирующий системный склероз (склеродермия).При этом заболевании чаще всего поражается кожа, хотя нередко страдают желудочно-кишечный тракт, почки, сердце, мышцы и легкие. У некоторых больных основным проявлением патологии длительное время остается поражение кожи, однако у большинства пациентов склеродермия прогрессирует в случае присоединения висцеральных проявлений. Смерть больных наступает от почечной, сердечной, легочной недостаточности или нарушения всасывания в тонкой кишке. Различают две разновидности течения заболевания:
▲ диффузную склеродермию, характеризующуюся широким вовлечением кожи, быстрым прогрессированием и ранними висцеральными проявлениями;
▲ местную склеродермию, сопровождающуюся относительно ограниченным вовлечением кожи (пальцы, предплечье, лицо). Висцеральные проявления присоединяются поздно, а течение заболевания относительно доброкачественное.
Этиология и патогенез.Прогрессирующий системный склероз — заболевание с неизвестной этиологией. Чрезмерное образование коллагена обусловлено взаимодействием многочисленных факторов, которые направлены на продукцию различных факторов роста фибробластов. В фиброгенезе играют роль как иммунологические, так и сосудистые нарушения (схема 32).
В соответствии с иммунологической гипотезой фиброз является следствием аномальной активации иммунной системы. Предполагают, что Т-лимфоциты, отвечая на какой-то неидентифицированный антиген, накапливаются в коже и выделяют цитокины, которые рекрутируют воспалительные клетки, включая тучные клетки и макрофаги. Некоторые медиаторы, продуцируемые тучными клетками и моноцитами, такие как гистамин, гепарин, ИЛ-1 и ФНО-а, могут усиливать рост фибробластов и увеличивать синтез коллагена. У многих больных склеродермией в коже находят активированные СО4+Т-лимфоциты-хелперы.
Все больные склеродермией имеют антинуклеарные антитела, которые реагируют с различными внутриядерными мишенями. Два типа антинуклеарных антител более или менее уникальны для прогрессирующего системного склероза. Один из них, направленный против топоизомеразы I ДНК, очень специфичен и присутствует у 28—70 % больных склеродермией. Больные, которые имеют антитела этого типа, чаще страдают легочным фиброзом и заболеваниями периферических сосудов. Антицентромерные антитела другого типа найдены у 22—36 % больных склеродермией и чаще встречаются у пациентов с ограниченным системным склерозом.
Сосудистая гипотеза основывается на наличии предшествующих сосудистых заболеваний у больных прогрессирующим системным склерозом. Фиброз внутренней оболочки пальцевых артерий, например, встречается у всех больных склеродермией. Отмечены также признаки повреждения эндотелия (повышенное содержание фактора Виллебранда) и активация тромбоцитов (увеличение количества циркулирующих тромбоцитов). Повторные повреждения эндотелия сопровождаются агрегацией тромбоцитов, что ведет к выбросу тромбоцитарных факторов, которые вызывают периадвентициальный фиброз. Активированные или поврежденные эндотелиальные клетки сами по себе могут выделять факторы, хемотаксические для фибро-бластов. Наконец, распространенное сужение сосудов микроцир-куляторного русла также приводит к ишемическому повреждению.
Схема 32. Патогенез прогрессирующего системного склероза (склеродермии)
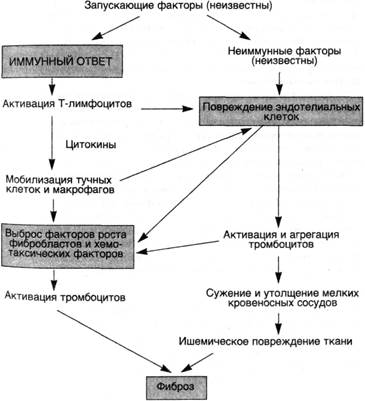
■ Таким образом, в основе прогрессирующего системного склероза лежат различные иммунные нарушения, выраженный фиброз и изменения микроциркуляторного русла. Хотя антигены, запускающие аутоиммунный ответ, и не идентифицированы, установлено, что именно иммунологические механизмы вызывают развитие фиброза с помощью цитокинов, которые активируют фибробласты, или посредством повреждения мелких кровеносных сосудов, либо благодаря обоим механизмам.
Воспалительные миопатии.Это гетерогенная группа заболеваний, характеризующихся иммунологически обусловленным воспалением скелетных мышц. К ним относятся дер-матомиозит и полимиозит, которые могут развиваться сами по себе или сочетаться с другими иммунологически обусловленными болезнями, обычно с прогрессирующим системным склерозом.
Дерматомиозит характеризуется поражением кожи и скелетных мышц, встречается у детей и взрослых. Классическая сыпь при этом заболевании возникает в виде сиреневых или обесцвеченных участков на верхних веках и сопровождается периорбитальным отеком. Нередко появляются шелушащиеся эритематозные высыпания или темно-красные пятна на суставах и локтях. Мышечная слабость развивается медленно, бывает двусторонней симметричной и обычно вначале поражает проксимальные мышцы, поэтому первыми симптомами заболевания бывают затруднения при вставании со стула и ходьбе вверх. Движения, контролируемые дистальными мышцами, страдают позже. Иногда, чаще у детей, возможны внемышечные проявления болезни в виде изъязвлений в желудочно-кишечном тракте и обызвествлений мягких тканей.
При полимиозите, так же как при дерматомиозите, поражаются симметричные проксимальные мышцы. Однако при полимиозите нет кожных проявлений. Он встречается главным образом у взрослых.
Этиология и патогенез.Этиология воспалительных миопатии неизвестна, но повреждение тканей, видимо, обусловлено иммунными механизмами.
При дерматомиозите основной мишенью служат капилляры. Микроциркуляторное русло атакуют антитела и компоненты комплемента, вызывая появление фокусов некроза миоцитов. При полимиозите, наоборот, возникают повреждения, опосредованные клетками. Около поврежденных мышечных волокон найдены CD8+ цитотоксические Т-лимфоциты и макрофаги, а экспрессия HLA-антигенов I класса увеличена на сарколемме нормальных мышечных волокон.
Как и при других аутоиммунных заболеваниях, при воспалительных миопатиях выявляются антитела.
Диагностика миозита основана на клинических симптомах, Данных электромиографии и биопсии.
Смешанные заболевания соединительной ткани.Описаны у тех больных, у которых сочетаются симптомы СКВ, полимиозита и прогрессирующего системного склероза, а серологически наблюдается высокий титр антител к рибонуклеопротеидам. При этих заболеваниях страдают почки; эффективно лечение кортикостероидами.
Для смешанных заболеваний соединительной ткани характерны артрит, опухание рук, феномен Рейно, аномальная подвижность пищевода, миозит, лейкопения и анемия, лихорадка, лимфаденопатия и гипергаммаглобулинемия.
Дата добавления: 2016-12-26; просмотров: 1177;