Нарушения фенотипического пола
Женский псевдогермафродитизм
Врожденная гиперплазия надпочечников. Клинические проявления. Пути образования глюкокортикоидов в надпочечниках и андрогенов в яичках и надпочечниках показаны на рис. 333-3. Три фермента принимают участие в синтезе как глюкокортикоидов, так и андрогенов (20,22-десмолаза, 3b-гидроксистероид-дегидрогеназан 17a-гидроксилаза); недостаточность любого из них препятствует образованию глюкокортикоидов и андрогенов и, следовательно, приводит как к врожденной гиперплазии надпочечников (из-за повышения уровня АКТГ), так и к недостаточной вирилизации эмбриона мужского пола (мужской псевдогермафродитизм). В синтезе андрогенов участвуют два фермента— 17,20-десмолаза и 17b-гидроксистероид-дегидрогеназа; недостаток какого-либо из них приводит к чистому мужскому гермафродитизму при нормальном синтезе глюкокортикоидов. Дефицит любого из двух последних ферментов синтеза глюкокортикоидов (21-гидроксилаза и 11b-гидроксилаза) нарушает образование гидрокортизона; компенсаторно возрастающая секреция АКТГ вызывает гиперплазию надпочечников и вторичное усиление выработки андрогенов, что приводит к вирилизации у женщин и преждевременной маскулинизации у мужчин.
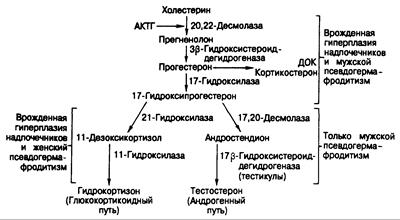
Рис. 333-3. Пути синтеза глюкокортикоидов и андрогенов.
Недостаточность надпочечников при этих нарушениях обусловливает тяжелую и угрожающую жизни патологию у лиц обоего пола. Подробно это рассматривается в гл. 325. Основные особенности разных форм врожденной гиперплазии надпочечников перечислены в табл. 333-4. Рассматривая нарушения полового развития, целесообразно проанализировать ферментные нарушения в стероидогенезе, приводящие либо к женскому, либо к мужскому псевдогермафродитизму. (Одно из таких нарушений — недостаточность 3b-гидроксистероид-дегидрогеназы — обусловливает как мужской, так и женский гермафродитизм, но, поскольку более распространенным пороком развития половых органов является неполная вирилизация у мужчин, эта ферментная патология рассматривается здесь как нарушение дифференцировки мужского фенотипа.)
Чаще всего причиной амбисексуальности половых органов у новорожденных служит врожденная гиперплазия надпочечников вследствие недостаточности 21-гидроксилазы (в Европе она встречается с частотой 1:5000, а в США — 1:15 000). Вирилизация у девочек проявляется обычно уже при рождении, а у мальчиков — в первые 2—3 года жизни. Для девочек характерны гипертрофия клитора в сочетании с его вентральным подтягиванием (патологическая эрекция), частичное сращение лабиоскотальных складок и вирилизация уретры различной степени. Внутренние женские половые органы и яичники остаются интактными, а вольфовы протоки регрессируют нормально, вероятно, потому, что надпочечники начинают функционировать на относительно поздних этапах эмбриогенеза. Наружные половые органы у девочек сходны с таковыми у мальчиков с двусторонним крипторхизмом и гипоспадией. Лабиоскротальные складки увеличены и морщинисты и напоминают мошонку. В редких случаях вирилизация достигает такой степени, что у девочки полностью развивается мужская уретра, половой член, а также предстательная железа, что приводит к ошибке при определении пола новорожденных. При рентгенографии после введения контрастного вещества в наружное половое отверстие обнаруживают влагалище, матку и иногда даже маточные трубы. В немногих случаях вирилизация девочек при рождении выражена незначительно или вообще отсутствует и проявляется лишь позднее — в детстве, отрочестве или зрелости. По-видимому, это связано с аллельной вариацией мутантных генов (так называемая поздно проявляющаяся, или взрослая, форма нарушения). Без лечения больные девочки в течение первого года жизни быстро растут и вирилизация у них прогрессирует. В период ожидаемого пубертата нормального полового созревания по женскому типу не происходит и менструации не появляются. Быстрое соматическое созревание у лиц обоего пола приводитк преждевременному заращению эпифизарных щелей и низкорослости в зрелые годы.
Таблица 333-4. Формы врожденной гиперплазии надпочечников
| Дефект | Кортизол | Альдостерон | Степень вирилизации у женщин | Степень недостаточной вирилизации у мужчин | Преимущественно секретируемый стероид | Примечания |
| Частичная недостаточность 21-гидр оксидазы (простая вирилизирующая или компенсирования форма) | Норма | | ++++ | 17-Гидроксипрогестерон | Самая распространенная форма (примерно 95% всех случаев); у 1/3—2/3 больных отмечается потеря соли | |
| Тяжелая недостаточность 21-гидроксплазы (сопр ов ождается потерей соли) | ¯ | ¯ | ++++ | 17-Гидроксипрогестерон | ||
| Недостаточность 1 lp-гид-роксилазы (гипертоническая форма) | ¯ | ¯ | ++++ | 11-Дезоксп-кортизоли 11-дезоксикорти-костерон | Гипертензия | |
| Недостаточность Зр-гпд-рокснстеропд-дегидрогеназы | о | + | ++++ | Д^Зр-ОН-сое-динения (дегидроэпиандростерон) | По-видимому, вторая по частоте встречаемости; обычно протекает с потерей соли | |
| Недостаточность 17a-гндроксидазы | ¯ | ¯ | ++++ | Кортикостерон и 11-дезоксикортикостерон | Отсутствие феминизации у женщин, гипертензия | |
| Недостаточность 20,22-десмолазы (липоидная гиперплазия надпочечников) | о | о | ++++ | Холестерин (?) | Редко встречающаяся форма; обычно протекает с потерей соли |
Так как дифференцировка мужского фенотипа остается нормальной, заболевание у мальчиков при рождении обычно не распознается, если только нет явной недостаточности надпочечников. Однако уже в первые годы жизни у больных наблюдают интенсивный рост и созревание наружных половых органов, частые эрекции и чрезмерное развитие мышц. Вирилизация у мальчиков может проявляться двояко. Избыточная секреция андрогенов надпочечниками ингибирует продукцию гонадотропинов, так что яички остаются незрелыми, несмотря на ускорение маскулинизации. В зрелые годы такие больные, если их не лечить, способны к эрекции и эякуляции, но сперматогенез у них отсутствует. В других случаях секреция андрогенов надпочечниками может активировать преждевременное созревание гипоталамо-гипо4шзарной оси и инициировать истинное преждевременное половое созревание, включая и сперматогенез (см. гл. 330). У нелеченых мужчин могут появляться АКТГ-зависимые «опухоли» яичек, состоящие из остатков клеток надпочечников.
При недостаточности 21-гидроксилазы, обусловливающей около 95% случаев врожденной гиперплазии надпочечников, продукция гидрокортизона уменьшается и, следовательно, возрастает секреция АКТГ, усиливается рост надпочечных желез, и тем самым происходит частичная или полная компенсация нарушения секреции гидрокортизона. Примерно у 50% больных отмечают частичную недостаточность фермента, и секреция кортизола остается нормальной. Эта форма заболевания называется простой вирилизирующей, или компенсированной. У остальных имеет место более полный дефицит фермента; даже увеличенные надпочечники не могут продуцировать адекватное количество кортизола и альдостерона, что приводит к выраженной потере соли с анорексией, рвотой, уменьшением объема жидкости и коллапсу в первые недели жизни. Это чак называемая форма недостаточности 21-гидроксилазы с потерей соли. У всех нелеченых больных отмечают избыточную продукцию предшественников кортизола, образующихся до стадии, катализируемой 21-гидроксплазой, в силу чего в плазме возрастает содержание прогестерона и 17-гидроксипрогестерона. Они действуют как слабые антагонисты альдостерона на рецепторном уровне, что при компенсированной форме требует большей, чем в норме, продукции альдостерона, чтобы сохранить нормальный баланс натрия.
Женский псевдогермафродитизм может быть вызван и недостатком 11b-гидроксилазы. В этом случае блокада гидроксилирования 11-го углеродного атома приводит к накоплению 11-дезоксикортизола и дезоксикортикостерона (ДОК) — сильного сользадерживающего гормона, что сопровождается не потерей соли, а гипертензией. Клинические проявления, обусловливаемые дефицитом глюкокортикоидов и избытком андрогенов, сходны с таковыми при недостаточности 21-гидроксилазы.
Патофизиология. Оба нарушения — следствие аутосомно-рецессивной мутации. Частота недостаточности 21 -гидроксилазы составляет примерно 1:50. Идентифицировано не менее трех форм недостаточности этого фермента, и все они связаны с мутациями генов, расположенных на 6-й хромосоме вблизи от локуса HLA-B: наиболее распространенный тип, проявляющийся как обычная аутосомно-рецессивная мутация с изменением ферментной активности: вариант, обусловленный криптическим аллелем, который даже у гомозигот не имеет никаких клинических проявлений, но вызывает типичное заболевание, если сосуществует с распространенным вариантом, и поздно проявляющийся вариант. Носители заболевания (равно как и гомозиготы) среди членов данной семьи могут быть идентифицированы но гаплотипу HLA. При недостаточности 11b-гндрокснлазы связь мутации с системой HLA остается неизвестной.
Эндокринная патология при этом состоянии рассматривается в гл. 325. Вкратце она сводится к повышению экскреции кетостероидов, как и основных метаболитов, накапливающихся выше места ферметной блокады У нелеченых больных повышено содержание АКТГ в плазме. При недостаточности 21-гидроксилазы в крови накапливается 17-гидроксипрогестерон, выводимый с мочой преимущественно в виде прегнантриола. При недостаточности 11-гидроксилазы в крови накапливается 11-дезокснкортизол, который выводится с мочой преимущественно в виде тетрагидрокортсксолона.
Лечение. Выбор пола должен определяться хромосомным и гонадиым полом, и соответствующую хирургическую коррекцию наружных половых органов следует производить как можно в более ранние сроки. Это весьма важно, так как при правильном лечении мужчины и женщины могут стать фертильными. Однако, если правильный диагноз устанавливается поздно (в возрасте старше 3 лет), выбор пола следует производить лишь после тщательного учета психосексуалыюй ориентации.
Консервативное лечение глюкокортикоидами предотвращает проявления недостаточности гидрокортизона, останавливает быструю вирилизацию и препятствует преждевременному соматическому развитию и заращению эпифизов. При недостаточности 11b-гидроксилазы подавление патологической секреции стероидов приводит к нормализации артериального давления, а при обоих вариантах обеспечивает своевременное начало менструальной функции и развитие женских вторичных половых признаков. У мужчин терапия глюкокортикоидами подавляет секрецию андрогенов надпочечниками и приводит к нормализации секреции гонадотропинов, развитию яичек и сперматогенезу. Контролируют заместительную терапию, определяя содержание в плазме 17-гидропрогсстерона, андростендиона. АКТГ и ренина. При тяжелых формах 21-гидроксилазной недостаточности, сопровождающейся потерей соли или повышением активности ренина в плазме, показано и лечение минералокортикоидами. У таких больных об адекватности заместительной минералокортикоидной терапии судят по активности ренина в плазме. Женский псевдогермафродитизм вненадпочечниковою генеза.Женский псевдогермафродитизм редко имеет вненадпочечниковые причины. В прошлом введение беременным женщинам для профилактики аборта прогестинов, обладающих побочными андрогенными эффектами (таких как 17a-этннил-19-нортестостерон), приводило к маскулинизации плодов женского пола. Женский псевдогермафродитизм может встречаться также у детей, рожденных матерями с вирилизирующими опухолями (например, арренобластомами или лютеомами беременных); в редких случаях причину заболевания установить не удается.
Врожденные дефекты мюллеровых протоков (врожденное отсутствие влагалища, агенезия мюллеровых структур). Клинические проявления. Врожденная гипоплазия, или отсутствие влагалища, в сочетании с аномалией или отсутствием матки (синдром Майера — Рокитанского — Кюстера — Хаузера) в качестве причины первичной аменореи уступает только дисгенезии гонад. У большинства больных нарушение диагностируют в возрасте ожидаемо! о полового созревания в связи с отсутствием менструаций. Рост и психическое развитие у них нормальны, а молочные железы, подмышечное и лобковое оволосение, а также телосложение соответствуют женскому типу. Матка может быть почти нормальной, лишенной только наружного входного канала, но чаще представлена рудиментарными двурогими тяжами с просветом или без него. У некоторых больных в области живота периодически появляются боли, что указывает на наличие достаточно функционального эндометрия, чтобы вызвать ретроградную менструацию и/или гематометру.
Примерно у 30% больных выявляют аномалии почек, чаще всего агенезию или эктопию. Встречается также сращение почек в виде подковы и солитарные эктопические почки, расположенные в тазовой полости. У 10% больных имеются нарушения скелета, причем у 60% из них в процесс вовлекается позвоночник, а у остальных — конечности и ребра. Специфические костные изменения характеризуются заклиниванием позвонков, их слиянием, рудиментарностью или асимметричностью тел позвонков и наличием дополнительных позвонков. Часто при этом наблюдают синдром Клнппеля—Фейля (врожденное сращение шейных позвонков, короткая шея, низкая задняя линия оволосения, а также болезненность и ограниченность движений шейного отдела позвоночника).
Патофизиология. Все больные имеют кариотип 46, XX. Чаще всего болезнь возникает спорадически, хотя наблюдали и несколько семейных случаев. Характер наследования в большинстве семейных случаев соответствует ограниченной полом аутосомно-доминантной мутации. Неясно, представляют ли собой спорадические случаи новые мутации того же типа, который определяет семейное нарушение, или они имеют многофакторную причину. Для семейных случаев характерна непостоянная экспрессируемость; у некоторых пораженных членов семьи имеются лишь скелетные или почечные аномалии, тогда как у других наблюдаются иные нарушения в производных мюллеровых протоков, например удвоение матки.
У мертворожденных плодов отсутствие матки и влагалища часто сочетается с двусторонней аплазией почек. Поэтому во всех случаях следует интересоваться наличием в семейном анамнезе изолированных нарушений скелета или ночек, а также мертворождений, которые могли бы быть связаны с врожденным отсутствием у плода обеих почек.
О сохранности функции яичников свидетельствуют овуляторные пики уровня ЛГ в плазме и двухфазные температурные кривые в течение цикла. У больных с нормальной маткой после хирургической пластики влагалища возможна беременность.
Лечение. Больных с агенезией влагалища можно лечить хирургически и консервативно. Цель хирургического вмешательства — создать искусственное влагалище путем имплантации резинового канала, покрытого несколькими слоями кожи. Консервативное лечение заключается в повторном давлении простым расширителем на влагалищную ямку, чтобы обеспечить ее достаточную глубину. Поскольку общая частота осложнений при хирургическом лечении составляет 5— 10%, у большинства больных следует пытаться использовать консервативный подход. Хирургическое же вмешательство можно рекомендовать женщинам с хорошо сформированной маткой, когда сохраняется возможность беременности. Чтобы сохранить новообразованное любым методом влагалище больной, целесообразно вести регулярную половую жизнь или проводить инструментальное расширение органа.
Мужской псевдогермафродитизм
Нарушение вирилизации эмбриона мужского пола (мужской псевдогермафродитизм) может быть следствием нарушения синтеза андрогенов или их действия, аномалий регрессии мюллеровых протоков и каких-то неясных причин. В 80% случаев мужского псевдогермафродитизма синтез андрогенов у больных остается нормальным.
Нарушения синтеза андрогенов. Клинические проявления. Известны пять ферментных дефектов, приводящих к нарушению синтеза тестостерона (см. рис. 330-3) и вызывающих неполную вирилизацию плода мужского пола в процессе эмбриогенеза. Все эти ферменты катализируют превращение холестерина в тестостерон на определенных этапах. Ферменты 20,22-десмолаза, 3b-гидроксистероид-дегидрогеназа и 17a-гидроксилаза принимают участие и в синтезе других гормонов надпочечников; поэтому их недостаток приводит не только к мужскому гермафродитизму, но и к врожденной гиперплазии надпочечников (см. табл. 333-4). Ферменты 17,20-десмолаза и 17b-гидроксистероид-дегидрогеназа участвуют только в синтезе андрогенов, и их недостаток приводит только к мужскому псевдогермафродитизму. Поскольку андрогены служат облигатными предшественниками эстрогенов, правильно заключить, что при всех таких нарушениях (кроме последнего этапа, катализируемого 17b-гидроксистероид -дегидрогеназой) у больных обоего пола будет снижен также синтез эстрогенов).
Нарушения функции надпочечников при трех соответствующих дефектах описаны в гл. 325, и здесь рассматриваются лишь нарушения полового развития. Улиц с кариотипом 46,XY матка и маточные трубы отсутствуют, что указывает на нормальную продукцию яичками в эмбриогенезе фактора, ингибирующего мюллеровы протоки. Маскулинизация вольфовых протоков, урогенитального синуса и урогенитального бугорка различна: у одних больных эти образования развиты нормально, у других полностью отсутствуют, поэтому клинические признаки соответствуют таковым у фенотипических мужчин с легкой гипоспадней или фенотипических женщин, которые до полного созревания напоминают больных с полной тестикулярной феминизацией. Эта крайняя вариабельность проявлений обусловлена разной степенью выраженности ферментных нарушений у разных больных и различным действием стероидов, накапливающихся проксимальнее мест метаболической блокады при разных нарушениях. У больных с частичными дефектами и у тех, у кого содержание тестостерона в плазме находится в пределах нормы, диагностировать заболевание можно лишь путем определения стероидов, накапливающихся выше места метаболической блокады.
Недостаточность 20,22-десмолазы (липоидная гиперплазия надпочечников) — это форма врожденной гиперплазии надпочечников, при которой в моче практически не удается обнаружить стероидов (ни 17-кетостероидов, ни 17-гидроксикортпкоидов). Нарушение затрагивает стадию, предшествующую образованию прегненолона, и, как предполагают, касается одного или нескольких ферментов 20,22-десмолазного комплекса, осуществляющего превращение холестерина в прегненолон. Синдром характеризуется потерей соли и выраженной недостаточностью надпочечников, большинство больных погибают в раннем детстве. При аутопсии находят увеличенные надпочечники и яички, инфильтрированные липидами. У больных мальчиков отмечается неполная маскулинизация, тогда как половые органы девочек развиваются нормально.
Недостаточность 3b-гндроксистероид-дегидрогеназы — вторая по распространенности причина врожденной гиперплазии надпочечников. У мальчиков она проявляется той или иной степенью гппоспадни или полным отсутствием маскулинизации вплоть до наличия влагалища. У новорожденных девочек отмечают признаки умеренной вирилизации из-за слабой андрогенной активностн дегидроэпиандростероиа — основного секретируемого стероида. Если фермент не вырабатывается ни в надпочечниках, ни я яичках, ни один из стероидов мочи не имеет d43-кето-конфигурацин, но у больных с частичным дефектом или поражением только яичек в моче обнаруживают нормальное или даже повышенное количество d43-кетостероидов. У большинства больных отмечают выраженную потерю соли и резкую недостаточность надпочечников. Больные с тяжелой недостаточностью фермента погибают. У больных мальчиков половое созревание протекает нормально, возможна лишь резко выраженная гинекомастия. В таких случаях уровень тестостерона в крови находится на нижней границе нормы, но концентрация d5-предшественников повышена. В разных тканях активность фермента регулируется по-разному, так как ферментная недостаточность в яичках может быть менее выраженной, чем в надпочечниках, а в печени фермент может полностью сохранить свою активность на фоне глубокой его недостаточности в надпочечниках и яичках. Отдифференцировать. лиц с нормальной активностью печеночных ферментов от больных с недостаточностью 21-гидроксилазы можно, лишь обнаружив, что содержание d5-прегнентриола в моче выше уровня прегнантриола.
Недостаточность 17a-гидроксилазы характеризуется гипогонадизмом, отсутствием вторичных половых признаков, гипокалиемическим алкалозом, гипертензией и практически полным выпадением секреции гидрокортизона при женском фенотипе. Секреция кортикостерона и дезоксикортикостерона (ДОК) надпочечниками повышена, а содержание 17-кетостероидов в моче понижено. Секреция альдостерона мала, что объясняется, по-видимому, высоким уровнем ДОК в плазме и снижением содержания ангиотензина. Однако после введения супрессивных доз гидрокортизона она нормализуется. Улиц с кариотипом 46,XX часто встречаются аменорея, отсутствие полового оволосения и гипертензия, но, поскольку для формирования женского фенотипа в эмбриогенезе половые стероиды не требуются, такие больные сохраняют нормальный фенотип препубертатных девочек. У мужчин, однако, недостаточность фермента приводит к нарушению вирилизация — от полного мужского псевдогермафродитизм а до амбисексуальности наружных половых органов с уретрой, открывающейся в промежности или мошонке. У мальчиков с частичной недостаточностью фермента в период полового созревания может развиться патологическая гинекомастия. Улиц с этим нарушением недостаточности надпочечников нет, так как у них повышена секреция кортикостерона (слабый глюкокортикоид) и ДОК (минералокортикоид). Гипертензия и гипокалиемия, которые являются яркими проявлениями этого нарушения (даже в неонатальном периоде), после подавления секреции ДОК соответствующими дозами глюкокортикоидов исчезают.
В некоторых семьях наблюдалась недостаточность 17,20-десмолазы. У больных мальчиков с набором хромосом 46,XY сохранялась нормальная функция коры надпочечников, но наблюдались те или иные признаки мужского псевдогермафродитизма. У большинства больных при рождении отмечали амбисексуальность наружных половых органов, но в период ожидаемого полового созревания происходит некоторая вирилизация. Однако у двух больных с кариотипом 46,XY наблюдали женский фенотип, вирилизации в возрасте ожидаемого полового созревания не происходило. Это нарушение отмечено также у одной женщины с кариотипом 4б,ХХ, страдающей половым инфантилизмом.
Недостаточность 17b-гндроксистероид-дегидрогеназы сказывается на последнем этапе биосинтеза андрогенов — восстановлении 17-кетогруппы андростендиона с образованием тестостерона. Это нарушение — наиболее частый дефект фер-
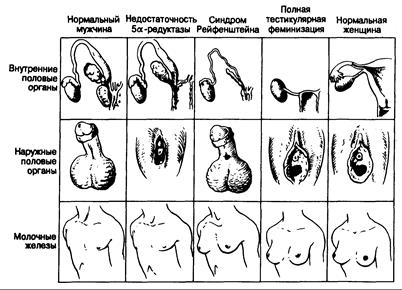
Рис. 333-4. Схематическое изображение внутренних и наружных половых органов, а также молочных желез при различных синдромах резистентности к андрогенам.
| Таблица 333-5. Анатомические, генетические и эндокринные особенности наследствен | |||||||||||
| ного мужского псевдогермафродитизма | |||||||||||
| Нарушение | Наследование | Фенотип | Эндокринная функция по отношению к | ||||||||
| здоровым мужчинам | |||||||||||
| мюллеровы | вольфовы | сперматогенез | урогенитальный синус | наружные | молочные | продукция | продукция | уровень Л1 | |||
| протоки | протоки | половые | железы | тестостерона | эстрогенов | ||||||
| органы | |||||||||||
| Нарушения синтеза тестостерона | |||||||||||
| Пять ферментных дефектов | Аутосомное или сцепленное с X-хромосомой рецессивное | Отсутствуют | Вариабельное развитие | Нормальный или сниженный | •i | Варьирует от мужского до женского | Как правило, женские | Обычно мужские | От нормальной до сниженной | Вариабельна | Повышен |
| t | |||||||||||
| Нарушения действия андрогенов | i | ||||||||||
| Недостаточность 5а-редуктазы Патология рецепторов: | Аутосомно-рецессивное | » | Мужские | Нормальный или сниженный | j | Женский | Клитороме-галия | Мужские | Нормальная | Нормальная | Нормален или повышен |
| Полная тестикулярная феминизация | Сцепленное с X-хромосомой рецессивное | » | Отсутствуют | Отсутствует | Женский | Женские | Женские | Повышена | Повышена | Повышен | |
| Неполная тестикулярная феми-минизация | Сцепленное с X-хромосомой рецессивное | » | Мужские | » | Женский | Клитороме-галия и заднее сращение | Женские | Повышена | Повышена | Понышен | |
| Синдром Реи-фенштейна | Сцепленное с X-хромосомой | » | Вариабельное развитие | » | Варьирует от мужского до женского | Неполное развитие по мужскому типу | Женские | Повышена | Повышена | 1 окышен | |
| Синдром мужского бесплодия | Вероятно, сцепленное с X-хромосомой рецессивное | » | Мужские | Отсутствует или сниженный | Мужской | Мужские | Обычно мужские | Нормальная или повышена | Нормальная или повышена | 1 h'pManb-iiblii 11 in повышен | |
| Резистентность при наличии рецепторов | Неизвестен | » | Вариабельны | Отсутствует или сниженный | Вариабельный | От женских до мужских | Вариабельны | Нормальная или повышена | Нормальная или повышена | Е-[орма;п,-ный или ич-вышеи | |
| Нарушения регрессии мюллеровых протоков | |||||||||||
| Синдром персистенции мюллеровых протоков | Аутосомное или сцепленное с X-хромосомой рецессивное | Рудиментарные матка и маточные трубы | Мужские | Нормальный | Мужской | Мужские | Мужские | Нормальная | Нормальная | Норм;!.11.- ный |
ментов синтеза тестостерона. Больные с мужским кариотипом 46,XY обычно имеют женский фенотип со слепо оканчивающимся влагалищем, производные мюллеровых протоков отсутствуют, но в паховой связке или брюшной полости находятся яички и вирилизированные структуры вольфовых протоков. Во время ожидаемого подового созревания происходит как вирилизация (с увеличением размеров полового члена и появлением волос на лице и туловище), так и выраженное в разной степени развитие молочных желез по женскому типу. У некоторых больных, если их не лечить, в пубертатном возрасте половое поведение меняется от женского к мужскому. Динамика андрогенов и эстрогенов в подробностях не изучена, но 17-кето-восстановление эстрона в эстрадиол в половых железах также снижено. Фермент 17b-гидроксистеронд-дегидрогеиаза в норме присутствует во многих тканях, кроме половых желез. Для данного же нарушения характерна его недостаточность, по-видимому, только в гонадах. Содержание тестостерона в плазме может находиться на нижней границе нормы, и поэтому для установления диагноза важно документировать повышение уровня андростендиона в плазме.
Патофизиология. Дефекты 17a-гидроксилазы и Зb-гидроксистероид-дегидрогеназы наследуются по аутосомно-рецессивному типу. Ограниченные данные о семейной распространенности дефицитов 17,20-дссмолазы и 17b-гндроксистероид-дегидрогеназы указывают либо на аутосомно-рецессивную, либо на сцепленную с Х-хромосомой рецессивную мутацию. Относительно же недостаточности 20,22-десмолазы имеющиеся данные не позволяют сделать определенного заключения о типе наследования.
Характер секреции и экскреции стероидов зависит от того, где локализуется та или иная метаболическая блокада (см. рис. 333-3). Как правило, секреция гонадотропинов повышена, и вследствие этого у многих больных с неполной недостаточностью ферментов последняя оказывается компенсированной, так что постоянная концентрация конечных продуктов, таких как тестостерон, может быть нормальной или близкой к норме.
В некоторых случаях мужского псевдогермафродитизма тестостерон образуется в недостаточном количестве не из-за дефицита какого-либо одного фермента синтеза андрогенов. К таким случаям относятся нарушения, где главным дефектом считают агенезию клеток Лейдига (возможно, вследствие отсутствия рецепторов ЛГ), или секрецию биологически неактивной молекулы ЛГ. Кроме того, как отмечалось выше, имеется ряд нарушении развития яичек, включая семейную XY-дисгснезию гонад, спорадическую дисгенезию яичек и синдром отсутствия яичек, при которых недостаточность продукции тестостерона оказывается вторичной по отношению к дефектам развития гонад.
Лечение. При нарушениях, сопровождающихся гипоплазией надпочечников, показана заместительная терапия глюкокортикоидами и в некоторых случаях — минералокортикоидами. Что касается аномалий половых органов, то решение об их коррекции следует принимать строго индивидуально. Больные с мужским гермафродитизмом бесплодны, что нужно принимать во внимание при выборе пола. У лиц с женским генотипом выбор пола не встречает трудностей (которые имеются при диагностике): больные воспитываются как женщины, в возрасте ожидаемого полового созревания им следует назначать заместительную терапию эстрогенами, чтобы индуцировать нормальное развитие женских вторичных половых признаков. Если же амбисексуальные половые органы находят у новорожденного мальчика, то решение вопроса о том, воспитывать ли его как мужчину или как женщину, зависит от анатомического дефекта; как правило, при более тяжелых нарушениях ребенка следует воспитывать как девочку и но возможности раньше производить хирургическую пластику половых органов и удаление яичек. Лицам, воспитанным в женском поле, в соответствующем возрасте также показана эстрогенная терапия, чтобы обеспечить нормальное развитие женских в торичных половых признаков. У лиц, воспитанных в мужском поле, следует производить хирургическую коррекцию любой имеющейся гипоспадии, а во время ожидаемого полового созревания строго следить за уровнем андрогенов и эстрогенов в плазме, чтобы определить необходимость хроническою дополнительного лечения тестостероном.
Нарушения действия андрогенов. Некоторые нарушения формирования мужского фенотипа обусловлены дефицитом действия андрогенов. Различные встречающиеся при этом фенотипы показаны на рис. 333-4 и охарактеризованы в табл. 333-5. При данной патологии образование тестостерона и регрессия мюллеровых протоков протекают нормально, но вследствие резистентности клеток-мишеней к действию андрогенов развитие по мужскому типу в тон или иной степени нарушается.
Недостаточность 5a-редуктазы. Эта аутосомно-рецессивная форма мужского псевдогермафродигизма характеризуется: 1) наличием у больных тяжелой промежностно-мошоночной гипоспадии с капюшонообразной крайней плотью, вентральной уретральной бороздкой и отверстием уретры в основании полового члена; 2) наличием слепого влагалищного кармана разных размеров, открывающегося либо в урогенитальный синус, либо на уретре кзади от ее отверстия; 3) наличием хорошо развитых яичек с нормальными придатками, семявыносящими протоками и семенными пузырьками, причем эякуляторные пути открываются в слепо заканчивающееся влагалище; 4) женским телосложением больных, не сопровождающимся развитием молочных желез но женскому типу; наличием нормального подмышечного и лобкового оволосения: 5) отсутствием женских внутренних половых органов; 6) наличием нормального для мужчин уровня тестостерона в плазме и 7) разной степенью маскулинизации больных в период полового созревания.
То обстоятельство, что нарушение вирилизации в процессе эмбриогенеза ограничивается урогенитальным синусом и закладкой наружных половых органов, позволяет понять природу главного дефекта. Тестостерон, секретируемый яичками плода, служит внутриклеточным медиатором дифференцировки вольфова протока в придаток яичка, семявыносящий проток и семенной пузырек, но вирилизация урогенитального синуса и наружных половых органов опосредуется дигидротестостероном. Следовательно, у эмбриона мужского пола с нормальным синтезом тестостерона и нормальными рецепторами андрогенов формирование фенотипа, свойственного индивиду при данном нарушении (нормальные производные вольфовых протоков с недостаточной маскулинизацией структур, образующихся из урогенитального синуса, полового бугорка и половых складок), следовало было бы ожидать при недостаточном образовании дигидротестостерона. Поскольку секрецияЛГ регулируется самим тестостероном (см. гл. 330), содержание этого гормона в плазме у таких больных повышено лишь незначительно. Поэтому скорости продукции тестостерона и эстрогенов остаются характерными для нормальных мужчин и гинекомастия не развивается.
Дефицит 5a-редуктазы при данном нарушении установлен с помощью непосредственного определения содержания этого фермента в биоптатах тканей и культурах фибробластов больных лиц. У большинства из них имеется либо резкий дефицит 5a-редуктазы, либо выпадение его функции, а у других ферментный белок, хотя и синтезируется с нормальной скоростью, но структурно отличается от нормального фермента. Остается неясным, почему вирилизация в пубертатном возрасте протекает активнее, чем та вирилизация, которая имеет место в процессе половой дифференцировки.
Патология рецепторов. Патология рецепторов андрогенов может приводить к формированию нескольких разных 4^енотипов. Несмотря на различия в клинической картине и молекулярных основах, эти нарушения имеют сходные эндокринологические, генетические и патофизиологические аспекты. Вначале будут рассмотрены главные клинические проявления патологии, а затем уже сходные особенности эндокринной 4)ункции и патогенеза.
Клинические проявления. Наиболее часто встречающейся формой псевдогермафродитизма является полная тестикулярная феминизация (от 1:20 000 до 1:64 000 новорожденных мальчиков). Она занимает третье по частоте место среди причин первичной аменореи улиц с женским 41енотипом после дисгенезии гонад и врожденного отсутствия влагалища. Женщины обращаются к врачу либо но поводу паховой грыжи (в препубертатном возрасте), либо по поводу аменореи (после полового созревания). Развитие молочных желез у больных, телосложение и распределение волос на теле и на волосистой части головы свойственны лицам женского пола, так что многие больные выглядят как настоящие женщины. Подмышечное и лобковое оволосение отсутствует или выражено слабо, но обычно имеется легкое оволосение вульвы. На лице растительности нет. Наружные половые органы женские, клитор нормального или несколько уменьшенного размера. Влагалище короткое и заканчивается слепо, но может вообще отсутствовать или находится в рудиментарном состоянии. Все внутренние половые органы отсутствуют. У больных обнаруживают лишь неопущенные яички, содержащие нормальные клетки Лейдига, и семенные канальцы; сперматогенеза нет.
Яички могут локализоваться в брюшной полости, по ходу пахового канала или в больших половых губах. Иногда в паратестикулярных фасциях или в фиброзных тяжах, идущих от яичек, присутствуют остатки мюллеровых или вольфовых структур. Больные, как правило, высокого роста, костный возраст и психическое развитие в пределах нормы. Психосексуальная ориентация в отношении поведения, внешнего вида и материнских инстинктов женская,
Основная опасность неопущения яичек, как и при других формах крипторхизма (см. гл. 330), кроется в опухолевом их перерождении. Поскольку у больных наблюдают нормальный пубертатный всплеск роста ц в возрасте ожидаемого полового созревания происходит феминизация и поскольку опухоли яичек при локализации последних в брюшной полости редко развиваются до постпубертатного возраста, кастрацию обычно откладывают до тех пор, пока не завершится срок ожидаемого полового созревания. Хирургическое вмешательство в препубертатном возрасте показано, если яички располагаются в паховой области или больших половых губах и создают дискомфорт или приводят к образованию грыжи. (При показаниях к грыжесечению в препубертатном возрасте большинство врачей предпочитают одновременно удалять яички, чтобы уменьшить число операций.) При удалении яичек в препубертатном возрасте необходимо своевременно начинать терапию эстрогенами, чтобы обеспечить нормальный рост и развитие молочных желез. Если же кастрацию производят в постпубертатном возрасте, то заместительная эстрогенная терапия поможет предотвратить появление симптомов менопаузы н других осложнений отмены эстрогенов (см. гл. 331).
Неполная тестикулярная феминизация встречается примерно в 10 раз реже, чем полная форма. В этих случаях незначительно выражена вирилизация наружных половых органов (частичное сращение лабиоскротальных складок и легкая клиторомегалия), нормальное оволосение лобка и некоторая вирилизация, равно как и феминизация во время ожидаемого полового созревания. Влагалище короткое и оканчивается слепо, но в отличие от полной формы патологии производные вольфовых протоков нередко бывают частично развитыми. Семейный анамнез обычно неинформативен, но в некоторых случаях патологию обнаруживают у многих членов семьи, причем характер наследования указывает на сцепленность признака с Х-хромосомой. Лечение больных с полной и неполной формами тестикулярной феминизации различно. Поскольку у больных с неполной формой в возрасте ожидаемого полового созревания происходит вирилизация, гонадэктомию при наличии клиторомегалии или заднего сращения половых губ следует производить в препубертатном возрасте.
Синдромом Ре и фен штейна называют различные формы неполного мужского псевдогермафродитизма. Ранее эти формы считали отдельными нозологическими единицами и называли по-разному — синдром Рейфенштейна, синдром Жильбера—Дрейфуса, синдром Лабса. Однако в настоящее время известны семьи, у больных членов которых проявления патологии варьируют, охватывая весь спектр фенотипов, описываемых этими терминами, и сейчас принято считать, что перечисленные синдромы представляют собой разные проявления единой мутации. Чаще всего болезнь характеризуется наличием промежностно-мошоночной гипоспадии и гинекомастии, но проявления нарушений вирилизации в пораженных семьях различны — от фенотипических мужчин с азооспермией до фенотипических женщин с псевдовлагалищем. Подмышечное и лобковое оволосение соответствует полу, но оволосение груди плица выражено в минимальной степени. Яички маленькие, часто наблюдаются крнпторхизм, азооспермия. У некоторых больных отмечают аномалии производных вольфовых протоков: например, у них отсутствуют или недоразвиты семявыносящие протоки. Поскольку психосексуальная ориентация больных в большинстве случаев безусловно мужская, гипоспадию и крнпторхизм следует корригировать хирургическим путем. Единственно успешный способ лечения при гинекомастии — хирургическое удаление молочных желез.
Синдром мужского бесплодия при патологии андрогенных рецепторов встречается чаще всего и в действительности не представляет собой какой-либо формы мужского псевдогермафродитизма. В некоторых случаях этот синдром является единственным проявлением семейного синдрома Рейфенштейна, а бесплодие у членов пораженной семьи обусловлено азооспермией вследствие рецепторных нарушений. Чаще же больные с мужским бесплодием не имеют семейного анамнеза; патология же рецепторов андрогенов может иметь место у 20% и более всех мужчин с идиопатической азооспермией. Эффективного лечения при любом из этих состояний нет.
Патофизиология. Кариотип у больных 46,XY, а мутантный ген сцеплен с Х-хромосомой. Семейный анамнез имеют примерно 60% больных с тестикулярной феминизацией и синдромом Рейфенштейна и отдельные больные с синдромом мужского бесплодия. Считают, что при отсутствии семейного анамнеза случаи заболевания обусловлены новыми мутациями.
Динамика гормонов при всех синдромах нарушения рецепторов андрогенов сходна. Содержание тестостерона в плазме и скорость его продукции яичками в пределах нормы или повышены. Повышение скорости продукции тестостерона обусловлено высокой средней концентрацией ЛГ в плазме, что в свою очередь объясняется нарушением механизма обратной связи вследствие резистентности гипоталамо-гипофизарной системы к действию андрогенов. Повышение содержания ЛГ определяет, вероятно, и увеличенную продукцию эстрогенов тестикулами (см. гл. 330). (У здоровых мужчин большая часть эстрогенов образуется путем периферического превращения андрогенов крови, но при повышении уровняЛГ в плазме значительное количество эстрогенов секретируется в кровь непосредственно яичками.) Таким образом, резистентность к регулирующему секрецию ЛГ действию андрогенов по механизму обратной связи приводит к повышению уровня ЛГ в плазме, а это в свою очередь обусловлнваег ускорение секреции как тестостерона, так и эстрадиола яичками. При удалении яичек содержание гонадотропинов возрастает еще больше, указывая на то, что секреция этих гормонов находится под частичным регуляторным контролем. Вероятно, в стабильных условиях и в отсутствие эффекта андрогенов секрецию ЛГ регулируют только эстрогены, что вызывает повышение концентрации эстрогенов в плазме крови у мужчин. Гормональные сдвиги при синдроме мужского бесплодия сходны с таковыми при других аномалиях рецепторов, по менее выражены. У некоторых больных с этим синдромом содержание ЛГ или тестостерона в плазме не повышается.
Феминизацию при описываемых нарушениях обусловливают два взаимосвязанных обстоятельства. Во-первых, андрогены и эстрогены на периферическом уровне обладают противоположными эффектами, и у здоровых мужчин вирилизация происходит при отношении андрогенов к эстрогенам 100:1 или выше; в отсутствие эффекта андрогенов действие эстрогенов на клетки не встречает сопротивления. Во-вторых, продукция эстрадиола превышает таковую у здоровых мужчин (хотя она и меньше, чем у здоровых женщин). Различная степень резистентности к андрогенам вместе с различной степенью повышения продукции эстрадиола и объясняет отличие признаков нарушения вирилизации и усиления феминизации при четырех клинических синдромах.
Любой из этих четырех синдромов связан с патологией рецепторов андрогенов. Вначале было показано, что в культурах фибробластов кожи некоторых больных с полной тестикулярной феминизацией почти совершенно отсутствует высокоаффинное связывание дигидротестостерона. Затем удалось установить, что у других больных с полной тестикулярной феминизацией, равно как у лиц с неполной тестикулярной феминизацией, синдромом Рейфенштейна и синдромом мужского бесплодия, имеют место либо снижение количества внешне нормальных рецепторов, либо качественные изменения рецепторов андрогенов.
Резистентность при наличии рецепторов. Разновидность резистентности к андрогенам, которая, по-видимому, не связана ни с дефицитом 5a-редуктазы, ни с нарушением рецепторов андрогенов, впервые была обнаружена в семье с синдромом тестикулярной феминизации. Затем были описаны больные с различными фенотипами — от неполной тестикулярной феминизации до синдрома Рейфенштейна. Гормональные сдвиги в этих случаях сходны с таковыми при патологии рецепторов. Природа молекулярного нарушения у таких больных остается неясной. Синдром мог бы быть связан с настолько тонкими аномалиями рецепторов андрогенов, что их не удается обнаружить обычными методами. Если же дефект действительно локализуется дистальнее рецептора, то он мог бы заключаться в неспособности клеток генерировать специфические информационные РНК или в нарушении процессинга РНК. На самом деле это заболевание может представить собой гетерогенную группу молекулярных нарушений. Лечение больных зависит от ихфенотипа.
Синдром персистенции мюллеровых протоков. Пораженные мужчины имеют нормальный половой член, но, кроме того, маточные трубы с обеих сторон, матку, верхнюю часть влагалища и по-разному развитые семявыносящие протоки. Больные часто обращаются к врачу по поводу паховой грыжи, в которой находится матка; нередко обнаруживается и крипторхизм. Семейный анамнез в большинстве случаев неинформативен, но описано несколько пар сиблингов, у которых данный синдром должен был бы наследоваться либо как аутосомно-рецессивная, либо как сцепленная с Х-хромосомой рецессивная мутация. Поскольку наружные половые органы хорошо развиты и в пубертатном возрасте происходит нормальная маскулинизация больных, полагают, что на критической стадии половой дифференцировки яички вырабатывают нужное количество андрогенов. Однако регрессии мюллеровых протоков не происходит, что можно объяснить неспособностью яичек плода продуцировать вещество, ингибирующее мюллеровы протоки, несвоевременной продукцией этого вещества или неспособностью тканей реагировать на этот гормон. Чтобы свести к минимуму опасность возникновения опухоли и сохранить вирилизацию, следует производить одномоментную или поэтапную орхиопексию. Злокачественные новообразования матки или влагалища не описаны, и, поскольку семявыносящие протоки тесно ассоциированы с широкими связками, матку и влагалище во время операции трогать не нужно, чтобы избежать травмы семявыносящих протоков и тем самым сохранить возможную фертильность.
Дефекты развития мужских половых органов. Гипоспадия. Гипоспадия —это врожденная аномалия, при которой уретра открывается по средней линии вентральной поверхности полового члена между нормальным расположением отверстия уретры и промежностью. Данный порок развития часто сопровождается той или иной степенью вентрального подтягивания и изгиба полового члена (патологическая эрекция); в США встречается у 0,5—0,8% новорожденных мальчиков.
Гипоспадию обычно подразделяют в зависимости от места расположения отверстия уретры — на головке полового члена, его теле или в промежностно-мошоночной области. Поскольку развитие полового члена опосредуется андрогенами, предполагают, что гипоспадия связана с каким-то ранним нарушением образования или действия андрогенов в процессе эмбриогенеза. Действительно, гипоспадия встречается при большинстве нарушений мужской половой дифференцировки. Иногда этот порок развития вызван приемом матерью на ранних стадиях беременности прогестиновых препаратов. В настоящее время известны причины (дефекты одиночного гена, хромосомные аномалии и прием матерью фармакологических средств) примерно 25% случаев гипоспадии, а причины большинства из них остаются неизвестными. Лечение хирургическое.
Крипторхизм. Нормальный процесс опущения яичек — это. вероятно, хуже всего изученный аспект мужской половой дифференцировки как в отношении природы сил, вызывающих перемещение яичек, так и гормональных факторов, регулирующих этот процесс. С анатомических позиций опущение яичек можно разделить на три стадии: 1) трансабдоминальное их перемещение от места образования над почками к паховому кольцу;
2) формирование отверстия в паховом канале (вагинальный отросток), через которое яички покидают брюшную полость; 3) прохождение яичек через паховый канал в мошонку. Весь этот процесс занимает более 6—7 мес беременности, начинаясь примерно на 6-й неделе и не заканчиваясь полностью у некоторых здоровых мальчиков даже к моменту рождения. Если андрогены и принимают участие в этом процессе, они, по-видимому, не являются единственными гормонами, обусловливающими нормальное опущение яичек. Если какой-либо из перечисленных выше процессов не произойди, это может привести к неопущению одного или обоих яичек (что встречается у 3% доношенных новорожденных мальчиков и у 30% недоношенных плодов мужского пола). Крипторхизм делится на интраабдоминальный, ретрактильный (периодическое втягивание яичек в паховый канал), обструктивный (постоянное расположение яичек в паху) и высокий мошоночный. У большинства больных отмечают ретрактильный крипторхизм, при котором в первые 6 нед — 3 мес жизни происходит постепенное опущение яичек, так что в позднем подростковом возрасте патология сохраняется лишь у 0,6—0,7% больных, которым и требуется искусственное низведение тестикул.
После полового созревания неопущенное яичко функционирует плохо, но неизвестно, до какой степени неопущение является результатом, а до какой — причиной нарушения тестикулярной функции. Предложены две основные теории возникновения крипторхизма — недостаточное интраабдоминальное давление и недостаточная эндокринная функция яичек в плане либо синтеза тестостерона, либо образования вещества, ингибирующего мюллеровы протоки. Действительно, врожденные пороки, приводящие к недостаточности интраабдоминального давления или развития самих яичек, могут сопровождаться крипторхизмом. Как и в случае гипоспадии, однако, известные причины крипторхизма лежат в основе лишь небольшой части случаев, а причины большинства остальных еще предстоит выяснить. Имеют значение два осложнения крипторхизма; при температуре брюшной полости сперматогенез не происходит, и поэтому, чтобы обеспечить возможную фертильность, коррекцию процесса необходимо осуществлять по возможности рано. Однако то обстоятельство, что мужчины, леченные по поводу как одностороннего, так и двустороннего крипторхизма, часто бесплодны, свидетельствует о том, что неопущение яичек обычно является следствием, а не причиной нарушения их функции. Отмечается также высокая частота злокачественного перерождения неопущенных яичек, и поэтому во всех этих случаях следовало бы прибегать к хирургическому вмешательству (см. гл. 297).
Дата добавления: 2016-03-05; просмотров: 1903;