Синдром Элерса—Данло
Общие проявления. Под названием «синдром Элерса—Данло» объединяют группу наследственных аномалий с повышенной подвижностью суставов и кожными проявлениями (рис. 319-5). Beighton вначале разделил этот синдром на пять типов (табл. 314-4). Тип I — это классическая тяжелая форма болезни, при которой отмечаются как чрезмерная подвижность суставов, так и типичная бархатистая и чрезмерно растяжимая кожа. Тип II сходен с I типом, но симптомы выражены слабее. При III типе чрезмерная подвижность суставов более выражена, чем изменения кожи. Тип IV характеризуется резким истончением кожи и частой внезапной смертью из-за разрыва крупных кровеносных сосудов или внутренних органов. Тип V сходен с типом II, но наследуется как сцепленный с Х-хромосомой признак.
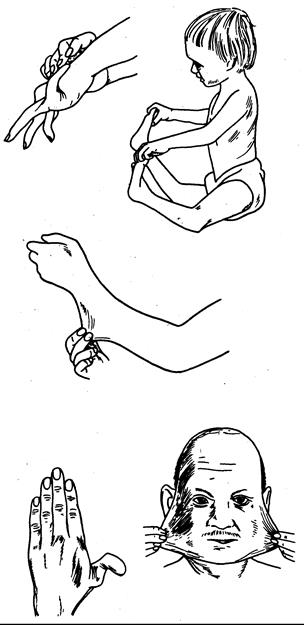
Рис. 319-5. Схематическое изображение кожных и суставных изменений при синдроме Элерса—Данло (СЭД).
Девочка (справа вверху) страдает СЭД IVB типа с дислокацией обоих бедер, не поддающейся хирургической коррекции [воспроизведено с разрешенияиз Prockop and Guzman, Hosp. Prac., 1977, 12(12):б1].
Таблица 319-4. Классификация больных с синдромом Элерса—Данло, основанная на клинических проявлениях и способе наследования
| Тип* | Чрезмерная подвижность суставов | Растяжимость кожи | Хрупкость | Склонность к кровоподтекам | Другие проявления | Тип наследования2 |
| I | Выражена | Выражена | Выражена | Выражена | Мягкая, бархатистая кожа; рубцы как папиросная бумага; грыжи; варикозно^расши-ренные вены; преждевременные роды из-за разрыва плодных оболочек | АД |
| II | Умеренная | Умеренная | Отсутствует | Умеренная | Менее выражены, чем при I типе | АД |
| III | Выражена | Минимально увеличена | Минимальная | Минимальная | Дислокация суставов с минимальными изменениями кожи | |
| IV | Только мелких суставов | То же | Выражена | Выражена | Разрыв крупных артерий и внутренних органов; тонкая кожа с выраженной венозной сетью; иногда характерные черты лица | АД или АР |
| V | Умеренная | Умеренная | Отсутствует | Умеренная | Сходные с таковыми при II типе | Х |
| VI | Минимально выражена | » | Умеренная | » | Сходные с таковыми при II типе; у некоторых больных внутримышечные кровоизлияния или кератоконус | Х |
| VII | Выражена | » | » | » | Множественные дислокации суставов | АР или АД |
| VIII | Умеренная | » | Выражена | » | Выраженный периодонтит; атрофические пигментированные рубцы на коже | АД |
| IX | Незначительно выражена | Незначительная | Отсутствует | Отсутствует | Дивертикулы мочевого пузыря со спонтанным разрывом; грыжи; костные аномалии; дряблость кожи | Х |
' Альтернативные названия: тип I — злокачественный, тип II — легкий, тип III — доброкачественная семейная чрезмерная подвижность суставов, тип IV — с кровоподтеками или аортальный, тип V — сцепленный с Х-хромосомой, тип VI — глазной, тип VII — врожденный множественный артрохалоз, тип VIII—периодонтальная форма, тип IX—синдром Элерса—Данло с нарушением метаболизма меди, синдром Менкеса (некоторые варианты) и дряблость кожи (некоторые варианты).
2 АД — аутосомное доминантное, АР — аутосомное рецессивное, Х — сцепленное с Х-хромосомой.
Впоследствии были выделены дополнительные типы (VI, VII и IX) с биохимическими нарушениями и фенотипами, не соответствующими типам, описанным Beighton. Однако не у всех больных с этими фенотипами были выявлены молекулярные дефекты, которые легли в основу классификации. Тип VII идентифицируется по генерализованному периодонтиту наряду с умеренными изменениями суставов и кожи. Многие больные и члены их семей не могут быть отнесены к больным ни одного из девяти упомянутых типов синдрома.
Изменения связок и суставов. Степень «разболтанности» и сверхподвижности суставов может варьировать от легкой до столь тяжелой, что сопровождается резкими невправимыми вывихами костей в тазобедренных и других суставах. При менее тяжелых формах больные могут сами вправлять вывихи или избегают их, ограничивая физическую активность. С возрастом у некоторых больных симптоматика усиливается, но в целом выраженная «разболтанность» суставов не уменьшает продолжительности жизни.
Кожа. Изменения кожи варьируют от некоторого ее истончения, мягкости и бархатистости до чрезмерной растяжимости и непрочности. Для больных с некоторыми типами синдрома характерны кровоподтеки. При IV типе через тонкую кожу просвечивают подкожные сосуды, при I типе при малейшей травме могут появляться полупрозрачные рубцы («папиросная бумага»). Сходные, но слабее выраженные признаки нарушенного заживления кожных травм имеются при других формах, особенно при V типе. У больных с VIII типом синдрома кожа отличается скорее хрупкостью, нежели растяжимостью, а раны на ней заживают, оставляя атрофические пигментированные рубцы.
Сопутствующие изменения. Помимо изменений суставов и кожи, у больных, особенно при I типе синдрома, может пролабировать митральный клапан сердца. Часто отмечаются плоскостопие и легкая степень или умеренно выраженный сколиоз. Выраженная «разболтанность» суставов с повторными вывихами может приводить к раннему остеоартриту. При I и IX типах нередко образуются грыжи, при IV типе могут быть спонтанные разрывы аорты и кишечника. При VI типе малейшие травмы глаз часто приводят к разрыву их оболочек, а кифосколиоз вызывает нарушение дыхания. При этом типе у больного склеры нередко имеют голубой цвет. При IX типе изменения суставов и кожи минимальны. Этот тип идентифицируется главным образом по нарушению обмена меди и включает состояния, ранее называвшиеся синдромом дряблости кожи (cutis laxa), наследуемым как признак, сцепленный с Х-хромосомой, сцепленным с Х-хромосомой синдромом Элерса—Данло и синдромом Менкеса. У больных часто образуются склонные к разрыву дивертикулы мочевого пузыря, грыжи и аномалии скелета, в том числе характерные затылочные «рога», а также дряблость кожи. При варианте, ранее обозначаемом как cutis laxa, именно дряблость кожи служит ведущим симптомом, придавая больным вид преждевременно состарившихся лиц. У них часто развиваются эмфизема легких и стеноз легочной артерии.
Молекулярные дефекты. При синдроме I, II и III типов молекулярные дефекты неизвестны. При электронной микроскопии кожи некоторых больных можно видеть необычное строение коллагеновых волокон, но аналогичные фибриллы иногда выявляют и в коже здорового человека.
У больных с IV типом болезни, по-видимому, имеется дефект синтеза или структуры коллагена III типа. Это согласуется с тем, что они склонны к спонтанным перфорациям аорты и кишечника, т. е. тканей, богатых коллагеном III типа. При одном из вариантов IV типа дефект заключается в синтезе структурно аномальных про-a(III)-цепей. Они входят в молекулу проколлагена III типа в равных стехиометрических соотношениях с нормальными про-a(III)-цепями, так что большинство молекул проколлагена III типа содержит одну или несколько аномальных про-a(III)-цепей. Эти молекулы подвергаются «самоубийству», или отрицательной комплементарности, и поэтому кожа практически не содержит коллагена III типа. При других вариантах IV типа нарушены синтез или секреция проколлагена III типа.
Синдром Элерса—Данло VI типа впервые был идентифицирован у двух сестер на том основании, что их коллаген содержал меньшее, чем в норме, количество гидроксилизина из-за недостаточности лизилгидроксилазы; недостаточность того же фермента была обнаружена и у других больных. Однако у некоторых больных с клинической картиной VI типа синдрома недостаточность лизилгидроксилазы не выявляется.
Синдром VII типа впервые был выделен как дефект превращения проколлагена в коллаген у больных с чрезмерной подвижностью суставов и вывихами. Это состояние на молекулярном уровне обусловлено двумя видами генетических нарушений. При одном из них (тип VIIA) имеется недостаточность проколлагена-протеиназы — фермента, отщепляющего N-концевой пептид от проколлагена I типа. Эта форма болезни наследуется как аутосомный рецессивный признак. Вторая форма (VIIБ) характеризуется рядом мутаций, придающих проколлагену I типа устойчивость к действию N-протеиназы. Для активности фермента необходима нативная конформация белкового субстрата, и на проколлаген I типа с измененной конформацией он не действует. Изменение аминокислотной последовательности в про-a-цепях проколлагена I типа может локализоваться на участке, отстоящем от места действия фермента на целых 90 аминокислот. При том и другом варианте (VIIA и VIIB) VII типа сохранение N-пропептида в молекуле приводит к образованию чрезвычайно тонких фибрилл. Как уже отмечалось, эти тонкие фибриллы могут участвовать в построении костей, но не обеспечивают необходимой прочности связкам и суставным сумкам.
У большинства обследованных больных с IX типом синдрома нарушен метаболизм меди (см. гл. 77). Низкий уровень меди и церулоплазмина в сыворотке сопровождается выраженным повышением уровня меди в клетках. Молекулярные дефекты у некоторых больных связаны, по-видимому, с синтезом диффундирующего фактора, принимающего участие в регуляции либо гена металлотионеина, либо каких-то других сторон метаболизма меди.
Диагностика. Диагностика все еще основывается на клинических признаках. Биохимические исследования для выявления известных нарушений до сих пор остаются очень трудоемкими и требующими больших затрат времени. При IV типе болезни инкубация культуры фибробластов кожи с радиоактивным пролином или глицином с последующим гель-электрофорезом новосинтезированных белков должна была бы обнаруживать нарушение синтеза или секреции проколлагена III типа. Для пренатальной диагностики этот подход в настоящее время неприменим. Исследование секреции и скорости процессинга проколлагена I типа в культуре фибробластов кожи дает в руки исследователей простой способ идентификации недостаточности проколлаген-N-протеиназы и структурных мутаций, препятствующих отщеплению N-концевого пропептида. Таким образом, этот способ мог бы оказаться полезным в диагностике VIIA и VIIБ вариантов VII типа синдрома. Однако положительные результаты анализа получают при обследовании и некоторых больных с несовершенным остеогенезом. При подозрении на синдром Элерса— Данло IX типа подтвердить диагноз можно путем определения уровня меди и церулоплазмина в сыворотке и культуре фибробластов. Вскоре можно ожидать применения анализа специфических ДНК при обследовании членов семей, у которых точно установлены генные мутации, характерные для синдрома I типа. Вероятно, в семьях с тяжелыми формами синдрома для пренатальной диагностики будет применяться и метод исследования полиморфизма длины рестрикционных фрагментов (см. также гл. 58).
Лечение. Специфического лечения не разработано. Хирургическая коррекция и укрепление суставных связок требуют тщательного индивидуального подхода, так как связки часто не держат швов. У всех больных, особенно при подозрении на IV тип, необходимо проверять состояние сердечно-сосудистой системы. При кровоподтеках определяют состояние свертывающей и антисвертывающей системы, но результаты этих исследований обычно не отличаются от нормы.
Синдром Марфана
Общие проявления. Синдром Марфана определяют по характерным изменениям трех видов соединительной ткани: скелета, глазной и сердечно-сосудистой (рис. 319-6). Синдром наследуется как аутосомный доминантный признак, причем 15—30 % его случаев приходится на свежие мутации. Относительно часто определяется «скачок через поколение», обусловленный непостоянной экспрессией. Кроме того, в некоторых семьях отдельные признаки (типичный «марфаноидный» вид, дислокация хрусталиков и нарушения кровообращения) могут наследоваться порознь. В связи с этим диагноз обычно не ставят до тех пор, пока хотя бы у одного члена семьи не выявят характерных изменений, по крайней мере в двух из трех соединительнотканных систем.
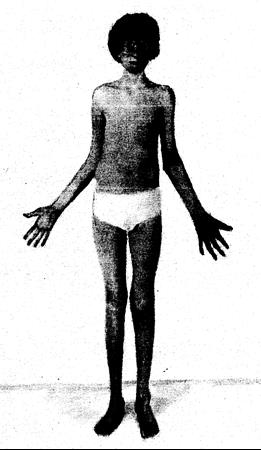
Рис. 319-6. Мальчик в возрасте 16 лет с синдромом Марфана. Проявления синдрома включают дислокацию хрусталиков глаз, удлиненное тонкое лицо, длинные пальцы рук (арахнодактилия), длинные конечности (долихостеномелия) и вдавление грудины (pectus excavatum) (любезно предоставлено J. G. Hall).
Аномалии скелета. Обычно рост больных выше, чем у родственников, руки и ноги у них заметно удлинены. Отношение верхней половины тела (от макушки до лобка) к нижней (от лобка до ступни), как правило, на два стандартных отклонения ниже среднего для соответствующих возраста, пола и расовой принадлежности. Пальцы рук и ног обычно длинные и тонкие (арахнодактилия или долихостеномелия), но объективно это трудно доказать. Из-за увеличения длины ребер грудная клетка часто деформируется, образуя вдавление («грудь сапожника») или выпячивание («куриная грудь»). Иногда грудная клетка явно симметрична. Обычно имеется сколиоз, часто с кифозом.
По подвижности суставов больных можно разделить на три группы. У большинства из них отмечается умеренная сверхподвижность многих суставов. У некоторых больных она выражена сильнее (как при синдроме Элерса—Данло), но у небольшого числа из них суставы тугоподвижны и имеются контрактуры рук и пальцев. Больные этой группы (контрактурная арахнодактилия), по-видимому, менее склонны к сердечно-сосудистым нарушениям.
Изменения сердечно-сосудистой системы. Обычно митральный клапан пролабирует, аорта расширена. Ее расширение начинается с корня и прогрессирует до расслаивающей аневризмы и разрыва. Для диагностики этих аномалий особенно полезна эхокардиография.
Глазные симптомы. Характерным признаком служит подвывих (эктопия) хрусталиков обычно по направлению вверх. Однако его можно обнаружить только при исследовании со щелевой лампой. Смещение хрусталиков в переднюю камеру глаза может вызвать глаукому, но она чаще развивается после удаления хрусталика. Длина оси глазного яблока больше нормы, что предрасполагает к близорукости и отслойке сетчатки.
Сопутствующие изменения. На коже плеча и ягодиц могут быть видны стрии. В остальном она остается неизмененной. У некоторых больных развивается спонтанный пневмоторакс. Часто имеют место высокие своды неба и стоп.
Диагностика. Легче всего установить диагноз, когда у больного или членов его семьи появляются объективные признаки подвывиха хрусталиков, расширения аорты и резкого кифосколиоза или деформаций грудной клетки. При эктопии хрусталиков и аневризме аорты диагноз ставят часто, даже если нет внешних «марфаноидных» признаков или семейного анамнеза. Всех больных с подозрением на этот синдром необходимо обследовать с помощью щелевой лампы и эхокардиографии. Следует также исключить гомоцистинурию (см. табл. 319-3) по отрицательным результатам цианиднитропруссидного теста на присутствие дисульфидов в моче. Эктопия хрусталиков может произойти и у больных с синдромом Элерса— Данло I, II и III типов, но у них отсутствует марфаноидный вид и определяются характерные изменения кожи, отсутствующие при синдроме Марфана.
Лечение.Как и при других наследственных болезнях соединительной ткани, определенных средств лечения при синдроме Марфана не существует. Некоторые специалисты рекомендуют использовать пропранолол (анаприлин) с целью предупредить тяжелые аортальные осложнения, но его эффективность не доказана. В ряде случаев проводилась хирургическая пластика аорты, аортального и митрального клапанов.
Сколиоз может прогрессировать, поэтому необходимы механическое укрепление скелета и физиотерапия, если он превышает 20°, или хирургическое, если он продолжает прогрессировать и превышает 45°. Для индукции менархе у девочек с прогрессирующим сколиозом применяли эстрогены, но определенных результатов получить не удалось.
Подвывих хрусталиков редко требует их удаления, но больные должны находиться под пристальным наблюдением из-за возможности отслоения сетчатки.
При консультировании исходят из 50 % вероятности наследования аномального гена. Из-за гетерогенности болезни ее выраженность у потомства может быть и большей, и меньшей, чем у родителей. Женщин следует информировать о высоком риске сердечно-сосудистых нарушений при беременности.
Дата добавления: 2016-03-05; просмотров: 1593;