Электролитическая диссоциация
Электролитомназывают вещество, которое в растворе или расплаве полностью или частично находится в форме заряженных частиц – ионов. Процесс распада молекул на положительно и отрицательно заряженные ионы – катионы и анионы – называется электролитической диссоциацией.
Длительное время электролитическая диссоциация рассматривалась как процесс, протекающий под воздействием электрического тока. Первая количественная теориярастворов электролитов была разработана С. Аррениусом в 1883 – 1887 гг. Согласно этой теории, электролитическая диссоциация происходит самопроизвольно при растворении веществ в соответствующих растворителях. В зависимости от природы электролита из одной молекулы могут образоваться два иона (например, при диссоциации NaCl, CuSO4), три (K2SO4, ZnCl2) или четыре (Na3PO4, AlCl3) иона. В зависимости от этого электролиты называют бинарными, тернарными, квартернарными.
Электролитической диссоциации может подвергаться лишь часть молекул вещества. Эта доля распавшихся молекул носит название степень диссоциацииa. Она определяется как отношение числа молекул n, распавшихся на ионы, к общему числу молекул N: a = n/N. Степень диссоциации зависит от природы растворенного вещества и растворителя, концентрации, температуры и может меняться в пределах 0 £ a £ 1. Вещества, полностью диссоциирующие на ионы, называются сильными электролитами, к слабым электролитам относят вещества, которые в умеренно концентрированных растворах находятся частично в ионной, частично в молекулярной формах. Следует иметь в виду, что эти понятия о сильных и слабых электролитах относятся не к самому веществу, а характеризуют его состояние в растворе, так как один и тот же электролит в зависимости от растворителя может диссоциировать полностью или в незначительной степени.
Степень диссоциации электролита зависит от концентрации, поэтому удобнее использовать в качестве меры силы электролита его константу диссоциацииK. Рассмотрим процесс диссоциации электролита ApBq , общая концентрация которого равна c, а степень диссоциации a:
ApBq L pAz+ +qBz-
(так как система в целом электронейтральна, то должно выполняться условие pz+ = qz- , где z+ и z- – заряды катиона и аниона).
При установившемся равновесии концентрация недиссоциированных молекул будет равна (1 – a)c, концентрация катионов c+ = pac, анионов c- = qac. Применяя к равновесию (15.1) закон действия масс, получим выражение для константы равновесия, являющейся в данном случае константой диссоциации:
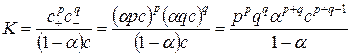 . (15.1)
. (15.1)
В случае бинарного 1-1 валентного электролита (p = q = 1; z+ = z–= 1) выражение (15.1) упрощается:
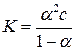 . (15.2)
. (15.2)
Если вместо концентрации использовать обратную ей величину – разведениеV = 1/c (т.е. объем, в котором содержится один моль растворенного вещества), то получим
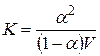 . (15.3)
. (15.3)
Уравнения (15.1) – (15.3) носят название закона разведения Оствальда.
Теория электролитической диссоциации Аррениуса позволила объяснить ряд известных к тому времени фактов, но не нашедших объяснения в рамках представлений о молекулярных растворах. Известно, что такие свойства растворов электролитов как понижение температуры замерзания, повышение температуры кипения растворителя в растворах, упругость пара над раствором, осмотическое давление отличаются от значений, вычисленных по общей концентрации растворенного вещества. Для учета этих отклонений Вант-Гофф ввел эмпирический поправочный множитель – изотонический коэффициентi, смысл которого становится совершенно ясным при использовании теории электролитической диссоциации, как величины, показывающей, во сколько раз увеличивается число частиц в растворе в результате диссоциации.
Если в растворе находится N молекул, каждая из которых диссоциирует на n ионов, а степень диссоциации равна a, то в растворе образуется anN ионов и остается (1 – a)N молекул, т.е. общее число частиц составляет anN + (1 – a)N. По сравнению с первоначальным числом N эта величина в i раз больше:
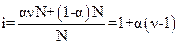 . (15.4)
. (15.4)
Теория Аррениуса объясняет также постоянство теплот нейтрализации сильных кислот сильными основаниями, так как в этих случаях реакции нейтрализации фактически сводятся к реакции между ионами
H+ + ОН– = Н2О
C позиций этой теории ясно, почему, например, при смешении водных растворов NaCl и KNO3 тепловой эффект практически равен нулю. Хотя формально в таком растворе возможна реакция
NaCl + KNO3 = KCl + NaNO3,
в действительности все вещества находятся в результате электролитической диссоциации в ионной форме, т.е. в растворе находится смесь ионов Na+, K+, Cl– и NO3–, которые химически не взаимодействуют друг с другом.
Однако по мере изучения свойств растворов электролитов выявились и недостатки теории Аррениуса. Так, например, оказалось, что степень диссоциации одного и того же электролита в одинаковых условиях, но определенная разными методами, не совпадает. В некоторых случаях степень диссоциации становится больше единицы, т.е. вообще лишается физического содержания. Константа диссоциации, вычисленная по закону разведения (15.4), которая по теории не должна зависеть от концентрации, остается постоянной только в очень разбавленных растворах слабых электролитов.
Кроме того, одним из главных недостатков теории являлось то, что она не указывала на причины электролитической диссоциации, на источники той большой энергии, которая необходима для разрыва связей в молекулах и переходу ионов в раствор.
Причиной этих недостатков являлось то, что частицы растворенного вещества уподоблялись молекулам идеального газа, находящимся в сплошной среде растворителя, и не рассматривалось взаимодействие частиц растворенного вещества друг с другом и с молекулами растворителя.
15.2. Сольватация ионов
По современным воззрениям, основы которых заложены в работах Каблукова (1891), энергия, которая обеспечивает разрыв связей в молекуле или кристаллической решетке, выделяется в результате взаимодействия растворяемого вещества с растворителем – процесса сольватации, а в случае водных растворов – гидратации.
Все вещества, которые при растворении образуют ионы, можно разбить на две группы. Первую группу составляют ионофоры– вещества, кристаллическая решетка которых состоит из ионов (NaCl, KNO3, Na2SO4 и т.п.). При сольватации происходит разрушение кристаллической решетки и образование свободных ионов.
К другой группе принадлежат ионогены– молекулярные вещества, ионы которых образуются лишь вследствие взаимодействия молекул с растворителем (HCl, CH3COOH и т.д.).
Но как для ионофоров, так и для ионогенов конечными продуктами их взаимодействия с растворителем являются сольватированные (гидратированные) ионы.
Энергии и теплоты гидратации ряда электролитов впервые были рассчитаны Борном и Габером с помощью циклов, основанных на законе Гесса. Эти расчеты и экспериментальные определения показали, что теплоты гидратации увеличиваются с возрастанием заряда ионов, а в ряду ионов с одинаковым зарядом – с уменьшением их радиуса, т.е. с увеличением плотности заряда.
Простейшая теориявзаимодействия ион – растворитель предложена Борном (1920). Согласно этой теории, ионы представляют собой сферы радиуса ri с зарядом zie (zi – зарядовое число иона, e – элементарный электрический заряд), а растворитель является сплошною средой с диэлектрической постоянной D. Изменение энергии Гиббса при сольватации DGs, по Борну, представляет разность энергий иона в растворе и в вакууме. Для расчета этой величины предлагается схема:
1) ион разряжается в вакууме; 2) нейтральная частица переносится в раствор; 3) частица в растворе заряжается до величины заряда иона.
Электрическая работа разряжения иона в вакууме
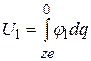 , (15.5)
, (15.5)
где j1 – электрический потенциал на поверхности иона:
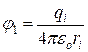 , (15.6)
, (15.6)
где qi – заряд иона, изменяющийся от zie до 0, eo – диэлектрическая проницаемость вакуума (eo = 8,85.10–12 Ф/м).
После подстановки уравнения (15.6) в (15.5) и интегрирования получим
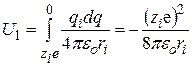 . (15.7)
. (15.7)
Незаряженную частицу переносят в растворитель с диэлектрической проницаемостью e = eoD, где D – диэлектрическая постоянная (относительная диэлектрическая проницаемость). В этом переносе никакой электрической работы не совершается. В растворителе частицу вновь заряжают до значения электрического потенциала на поверхности
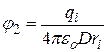 . (15.8)
. (15.8)
Электрическая работа заряжения равна
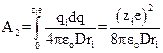 (15.9)
(15.9)
Суммарная работа переноса 1 моль ионов из вакуума в раствор при постоянных давлении и температуре представляет собой энергию сольватации
DGs = – NA (A1 + A2),
откуда получаем уравнение Борна для энергии сольватации ионов:
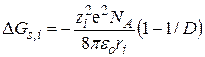 . (15.10)
. (15.10)
Дифференцированием этого уравнения по температуре получают энтропию сольватации DSs,i , а затем и энтальпию сольватации DHs,i:
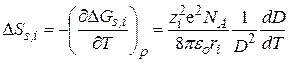 ; (15.11)
; (15.11)
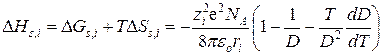 . (15.12)
. (15.12)
Теория Борна является приближенной, и рассчитанные по уравнению (15.10) энергии сольватации лишь качественно правильно отражают зависимости от заряда и радиуса ионов. В ряде случаев не соблюдается также линейная зависимость энергии сольватации от обратной диэлектрической постоянной. Это связано с тем, что в действительности вблизи иона вследствие диэлектрического насыщения диэлектрическая проницаемость отличается от ее значения в чистом растворителе. Кроме того, растворитель не является сплошной средой и обладает некоторой структурой, которая изменяется под действием ионов, которые при сольватации в большей или меньшей степени связывают молекулы растворителя. Лучше всего эти процессы исследованы в водных растворах.
 Рис. 15.1. Структура жидкой воды (а) и ее изменения под влиянием катиона (б)
Рис. 15.1. Структура жидкой воды (а) и ее изменения под влиянием катиона (б)
|
Вода относится к ассоциированным жидкостям, т.е. к таким, у которых силы, действующие между молекулами, имеют определенное направление в пространстве и локализованы в определенных частях молекулы. Чистая вода при 0o С имеет ажурную тетраэдрическую структуру, обусловленную наличием направленных водородных связей, энергия которых составляет около 20 кДж/моль. Координационное число, т.е. число ближайших соседей около данной молекулы, для жидкой воды при 0o С равно 4. При повышении температуры координационное число увеличивается, что связывается с частичным разрушением квазикристаллической решетки и попаданием свободных молекул в полости структуры воды.
Если в тетраэдрической структуре жидкой воды одну молекулу заменить частицей, обладающей большим зарядом, например, катионом, то структура должна измениться. Две молекулы воды должны еще прочнее закрепиться на своем месте, а две другие – повернуться своими отрицательными полюсами к катиону (молекулы 1, 4 на рис. 15.1 б). В зависимости от величины заряда и размеров иона первоначальное расположение молекул воды может сохраниться или же может возникнуть новая жесткая структура, возможны также различные промежуточные состояния. Некоторые ионы могут нарушать первоначальное упорядоченное состояние, но не способствовать возникновению новой структуры; такое явление называют эффектом деструктурирования.
О.Я.Самойлов предложил рассматривать гидратацию как изменение трансляционного движения молекул воды, находящихся вблизи иона. Если молекула воды находится рядом с другой такой молекулой время t, а рядом с ионом – ti , то мерой гидратации является отношение t/ti. При t/ti < 1 молекулы воды прочно удерживаются ионом ("положительная гидратация"), если же t/ti > 1 молекулы около иона обмениваются легче, чем в чистом растворителе, ион разрушает структуру воды ("отрицательная гидратация").
Учет структуры растворителя, а также различных сил взаимодействия, кроме электростатических, привели к развитию методов расчета энергий сольватации, которые дают лучшие результаты, чем уравнение Борна. Однако в силу сложности и многообразия сил взаимодействия в растворах до сих пор нет достаточно надежных и точных теоретических методов расчета энергии сольватации ионов в различных растворителях.
15.3. Активности и коэффициенты активности электролитов
Отклонения свойств растворов электролитов от идеальных обусловлены взаимодействием между частицами в растворах. Эти взаимодействия для ионов за счет электростатических кулоновских сил проявляются в большей степени и на больших расстояниях, чем силы ван-дер-ваальсовского взаимодействия в молекулярных растворах, и вызывают отклонения от идеальности даже в разбавленных растворах.
В связи с этим для описания свойств растворов электролитов вместо концентраций используются активности. Активность ионовопределяется так же, как и активность молекул неэлектролитов. В качестве стандартного обычно выбирается состояние, в котором активность и коэффициент активности равны единице. Так для моляльного коэффициента активности используется соотношение:
ai = gimi; 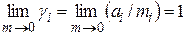 . (15.13)
. (15.13)
В силу условия электронейтральности термодинамические свойства отдельного сорта ионов не могут быть определены, поэтому при описании свойств электролитов используются средние ионные активности a±и средние ионные коэффициенты активности g±.
Рассмотрим раствор, состоящий из n1 молей растворителя и n2 молей растворенного вещества. Согласно уравнению Гиббса – Дюгема
n1dlna1 + n2dlna2 = 0, (15.14)
где a1 и a2 – активности растворителя и растворенного вещества. Если взять 1000 г растворителя, то n1 = 1000/M1 (M1 – молярная масса растворителя), а моляльность m = n2; тогда
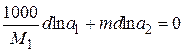 . (15.15)
. (15.15)
Растворенное вещество диссоциирует на ионы с концентрацией катионов m+ и анионов m–, и можно записать:
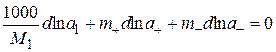 . (15.16)
. (15.16)
В уравнениях (15.15) и (15.16) активность растворителя a1 представляет одну и ту же величину. Вычитая (15.15) из (15.16), получим:
m+dlna+ + m–dlna– – mdlna2 = 0. (15.17)
В случае полной диссоциации растворенного электролита концентрация катионов m+ = n+m и анионов m– = n–m. Подставив эти значения в уравнение (15.16), получим:
n+dlna+ + n–dlna– – dlna2 = 0 (15.18)
или
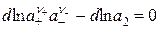 . (15.19)
. (15.19)
Таким образом, в уравнении (15.19) мы получим не активности отдельных ионов, а произведение 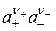 .
.
Из уравнения (15.21) следует, что
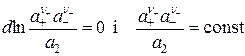 . (15.20)
. (15.20)
Из изложенного видно, что активность растворенного электролита можно представить в форме общей активности a2, без учета диссоциации, или через ионные активности a+ и a–. Если выбрать для a2 стандартное состояние так, чтобы постоянная в уравнении (15.22) равнялась единице, то связь между общей активностью и активностями отдельных ионов будет представлена в виде
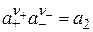 . (15.21)
. (15.21)
Так как a+ и a– определить в отдельности строго термодинамически невозможно, то используют среднюю ионную активность a±:
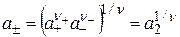 . (15.22)
. (15.22)
Аналогично вводятся понятия о средней ионной моляльности и среднем ионном коэффициенте активности:
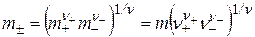 , (15.23)
, (15.23)
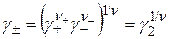 . (15.24)
. (15.24)
Связь между средними ионными активностью, моляльностью и коэффициентами активности устанавливается соотношением:
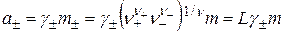 , (15.25)
, (15.25)
где 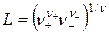 зависит от зарядного типа электролита.
зависит от зарядного типа электролита.
Аналогичные соотношения получаются при выражении состава раствора не через моляльности, а в других шкалах – молярности, мольной доли, однако численные значения активностей и коэффициентов активности будут, конечно, другими. Количественные соотношения между ними можно получить, исходя из значений химического потенциала, величина которого не зависит от выбора концентрационной шкалы:
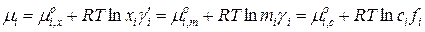 , (15.26)
, (15.26)
где, 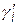 , gi и fi – рациональный, моляльный (практический) и молярный коэффициенты активности ионов i-го сорта. Стандартные состояния во всех шкалах выбираются таким образом, что в бесконечно разбавленном растворе все три коэффициента активности равны единице, а концентрации связаны между собой соотношениями:
, gi и fi – рациональный, моляльный (практический) и молярный коэффициенты активности ионов i-го сорта. Стандартные состояния во всех шкалах выбираются таким образом, что в бесконечно разбавленном растворе все три коэффициента активности равны единице, а концентрации связаны между собой соотношениями:
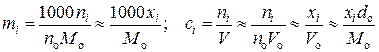 , (15.27)
, (15.27)
где no, Mo, Vo, do – число молей, молярная масса, молярный объем и плотность растворителя, а V – объем раствора. Тогда стандартные химические потенциалы связаны между собой соотношением:
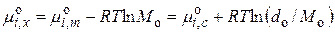 . (15.28)
. (15.28)
Подставив значения стандартных потенциалов в уравнение (15.28), получим для ионного коэффициента активности:
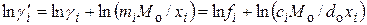 . (15.29)
. (15.29)
Для средних ионных коэффициентов активности при любых концентрациях получаем:
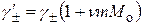 ;
;
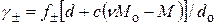 ;
;
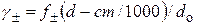 , (15.30)
, (15.30)
где d и M – плотность раствора и молярная масса растворенного вещества.
В растворе одновременно может находиться несколько электролитов, ионы которых оказывают взаимное влияние на поведение друг друга, и коэффициент активности каждого электролита будет зависеть не только от его концентрации, но и от концентрации других электролитов. Поэтому для характеристики общей концентрации ионов было введено понятие ионной силыраствора I, которая равна полусумме произведений концентрации каждого иона на квадрат его заряда:
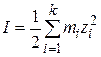 . (15.31)
. (15.31)
Многочисленные исследования показали, что в разбавленных растворах коэффициенты активности электролитов одинакового валентного типа при данной ионной силе совпадают. Эти результаты были обобщены Льюисом и Рендаллом и сформулированы в виде правила ионной силы:
В растворе с данной ионной силой все электролиты имеют значения коэффициентов активности, не зависящие от природы и концентрации вещества, а зависящие лишь от числа и заряда его ионов.
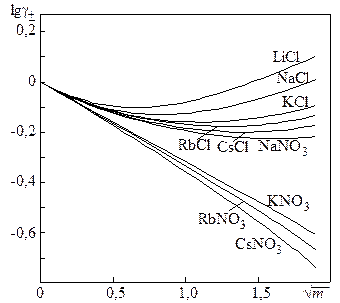 Рис. 15.2. Зависимость логарифма коэффициентов активности некоторых 1 – 1 электролитов от корня из ионной силы
Рис. 15.2. Зависимость логарифма коэффициентов активности некоторых 1 – 1 электролитов от корня из ионной силы
|
Если представить зависимость коэффициентов активности от ионной силы в форме lgg± – 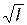 , то в области разбавленных растворов получаются прямые, наклон которых одинаков для всех электролитов одного зарядного типа, т.е. получаем, что lgg± = – A , где коэффициент A зависит только от зарядного типа электролита (рис.15.1).
, то в области разбавленных растворов получаются прямые, наклон которых одинаков для всех электролитов одного зарядного типа, т.е. получаем, что lgg± = – A , где коэффициент A зависит только от зарядного типа электролита (рис.15.1).
Однако эта зависимость точно выполняется лишь при очень малых ионных силах (I £ 0,01 – 0,02). Теоретическое обоснование правила ионной силы было дано Дебаем и Гюккелем в разработанной ими теории растворов сильных электролитов.
15.4. Теория растворов сильных электролитов
Термодинамический потенциал G реального раствора отличается от термодинамического потенциала Gид идеального раствора на некоторую величину G*, обусловленную межионным взаимодействием:
G = Gид + G* . (15.32)
Химический потенциал i-го сорта ионов в реальном растворе
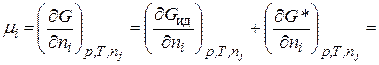
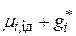 . (15.33)
. (15.33)
Выражая концентрацию числом ионов в единице объема, для химических потенциалов ионов в реальном и идеальном растворах запишем:
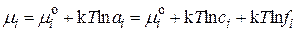 ; (15.34)
; (15.34)
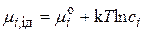 . (15.35)
. (15.35)
Сравнивая эти уравнения и учитывая (15.33), получим:
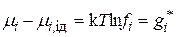 . (15.36)
. (15.36)
Таким образом, величина коэффициентов активности ионов определяется энергией межионного взаимодействия 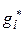 .
.
Точное определение этой величины в настоящее время невозможно вследствие недостаточности нашего знания о строении растворов и природе сил, действующих в растворе. Приближенное решение задачи можно получить на основе какой-либо модели раствора, отражающей его основные характеристики.
Основные положения теории растворов электролитов были даны в работах Дебая и Гюккеля(1923), которые исходили из следующих представлений о растворе:
1. Предполагается, что электролит полностью диссоциирован в растворе, т.е. теория применима к растворам сильных электролитов.
 Рис. 15.3. Модель ионной атмосферы Рис. 15.3. Модель ионной атмосферы
|
2. Распределение ионов в растворе определяется двумя факторами. С одной стороны, благодаря электростатическому межионному взаимодействию, в растворе сохраняется распределение, подобное распределению ионов в кристаллической решетке, где каждый ион окружен преимущественно ионами противоположного знака заряда. С другой стороны, такое распределение нарушается тепловым движением ионов. В результате вокруг любого иона, выбранного в качестве центрального, образуется ионная атмосфера, состоящая как из ионов того же знака заряда, что и у центрального, так и противоположного. Любой центральный ион входит в то же время в состав ионной атмосферы другого иона, а в результате теплового движения все ионы перемещаются, меняются местами, выходят из ионной атмосферы и входят в ионную атмосферу другого иона (рис. 15.3). Таким образом, представления об ионной атмосфере являются некоторым усредненным, статистическим понятием. Размер ионной атмосферы выбирается таким, что ее заряд оказывается равным по величине и противоположным по знаку заряду центрального иона, т.е. зависит от концентрации электролита.
3. В качестве энергии взаимодействия рассматривается только электростатическая энергия притяжения и отталкивания ионов, причем рассчитывается энергия взаимодействия центрального иона с ионной атмосферой в целом, без учета дискретного распределения зарядов, а ион рассматривается как точечный заряд.
4. Растворитель представляется в виде непрерывной среды с диэлектрической проницаемостью e, поэтому взаимодействие ионов с молекулами растворителя (сольватация) не учитывается.
Электрическая энергия ионной атмосферы зависит от плотности электрического заряда и потенциала ионной атмосферы. Связь между ними можно выразить с помощью уравнения Пуассона:
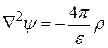 , (15.37)
, (15.37)
где y – средняя величина потенциала в данной точке,
r – средняя объемная плотность заряда,
e – диэлектрическая проницаемость,
Ñ2 – оператор Лапласа, который для прямоугольных координат равен
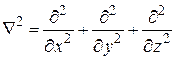 .
.
Для нахождения плотности заряда рассмотрим раствор, в единице объема которого (например, в 1 см3) находится по ni ионов i-го сорта с зарядом zi. Для такого объема раствора выполняется закон электронейтральности, т.е.
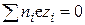 или
или 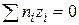 . (15.38)
. (15.38)
 Рис. 15.4. Распределение зарядов в поле центрального иона
Рис. 15.4. Распределение зарядов в поле центрального иона
|
Однако, в достаточно малом элементе объема dV, находящемся на расстоянии r от иона, выбранного в качестве центрального, вследствие существования ионной атмосферы заряд будет отличаться от нуля (рис. 15.4). В этом элементе объема dV находится по dni ионов каждого вида.
Если предположить, что к распределению ионов приложим закон распределения Больцмана, то число ионов каждого сорта в этом объеме
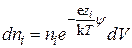 , (15.39)
, (15.39)
а заряд элемента объема, создаваемый всеми ионами, равен
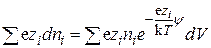 , (15.40)
, (15.40)
и средняя плотность заряда, т.е. величина заряда единицы объема равна
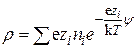 . (15.41)
. (15.41)
Подставляя это значение в уравнение Пуассона (15.37), получим:
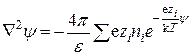 . (15.42)
. (15.42)
В связи с тем, что уравнение (15.42) не имеет решения в общем виде, авторы теории предположили, что eziy<<kT, т.е. что электростатическое взаимодействие значительно слабее энергии теплового движения. Это условие реально может выполняться лишь в достаточно разбавленных растворах. В этом случае показательную функцию можно разложить в ряд, ограничившись только двумя первыми членами разложения. Тогда для плотности заряда получим:
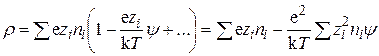 . (15.43)
. (15.43)
Первый член суммы справа в этом уравнении по условию электронейтральности (15.38) равен нулю, и уравнение (15.42) можно записать в виде
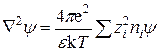 . (15.44)
. (15.44)
Так как ионная атмосфера имеет шаровую симметрию, удобно перейти от прямоугольной к сферической системе координат, в которой
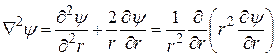 , (15.45)
, (15.45)
где r – расстояние от центрального иона до точки, в которой определяется потенциал y.
Введем обозначение
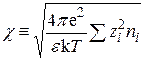 . (15.46)
. (15.46)
Подставив уравнения (15.45) и (15.46) в уравнение (15.44), получим дифференциальное уравнение:
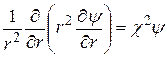 , (15.47)
, (15.47)
общее решение которого дает
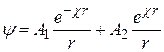 . (15.48)
. (15.48)
Для нахождения коэффициентов A1 и A2 используем граничные условия. При удалении от центрального иона потенциал уменьшается и в пределе при r ® ¥ потенциал y ® 0. Это условие выполняется лишь при A2 = 0. С другой стороны, при приближении к иону (r ® 0) потенциал y должен стремиться к потенциалу самого иона yi , т.е.
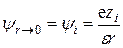 . (15.49)
. (15.49)
Разложим экспоненту e–cr в ряд:
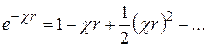 . (15.50)
. (15.50)
При r ® 0 все члены разложения значительно меньше единицы и ими можно пренебречь. Тогда из уравнения (15.48), учитывая, что A2 = 0, получим:
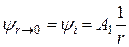 . (15.51)
. (15.51)
Из сравнения уравнений (15.49) и (15.51) следует, что
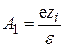 , (15.52)
, (15.52)
и уравнение (15.50) приводится к виду
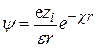 . (15.53)
. (15.53)
Общая величина потенциала y обусловлена значениями потенциала, создаваемого ионной атмосферой ya и центральным ионом yi в данной точке ионной атмосферы. Потенциал, создаваемый ионной атмосферой на расстоянии r от центрального иона
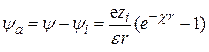 . (15.54)
. (15.54)
Энергию взаимодействия можно рассматривать как потенциал ионной атмосферы в точке нахождения центрального иона. Разлагая e–cr в ряд и ограничившись двумя первыми членами разложения, получим
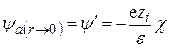 . (15.55)
. (15.55)
Сравнивая это выражение с уравнением (15.49), видим, что величину y¢ можно рассматривать в качестве потенциала, создаваемого некоторым ионом, находящимся на расстоянии 1/c от центрального и с противоположным знаком заряда, в точке нахождения этого центрального иона. Фактически же потенциал создается ионной атмосферой, поэтому величина 1/c, называемая характеристической длиной, представляет собой радиус ионной атмосферы, окружающей центральный ион.
Электростатическую энергию межионного взаимодействия 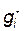 можно представить как работу заряжения центрального иона от нуля до заряда q в поле ионной атмосферы:
можно представить как работу заряжения центрального иона от нуля до заряда q в поле ионной атмосферы:
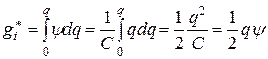 , (15.56)
, (15.56)
где C – электрическая емкость и q = Cy.
Так как заряд иона qi = ezi, а потенциал ионной атмосферы, создаваемый в точке нахождения центрального иона, определяется уравнением (15.54), то
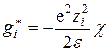 . (15.57)
. (15.57)
Это основное уравнение теории Дебая – Гюккеля, позволяющее рассчитывать различные свойства растворов электролитов.
15.5. Расчет коэффициентов активности ионов
Одним из важных следствий, вытекающих из основного уравнения, является возможность расчета коэффициентов активности ионов. Учитывая связь энергии межионного взаимодействия с коэффициентами активности (уравнение (15.36)), получаем:
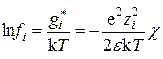 . (15.58)
. (15.58)
Подставим сюда значение c из уравнения (15.46):
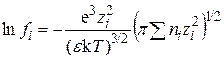 . (15.59)
. (15.59)
В этом уравнении концентрация выражена числом ионов ni в 1 см3 раствора. Эта величина связана с молярной концентрацией ci: 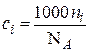 , (где NА – число Авогадро), тогда
, (где NА – число Авогадро), тогда
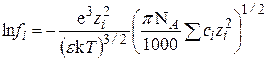 . (15.60)
. (15.60)
Преобразуем это уравнение, вводя в него ионную силу 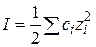 , переходя к десятичным логарифмам и обозначив
, переходя к десятичным логарифмам и обозначив
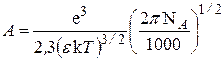 . (15.61)
. (15.61)
После этих подстановок уравнение для коэффициентов активности ионов принимает вид:
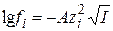 . (15.62)
. (15.62)
Уравнение Дебая – Гюккеля(15.62) дает возможность рассчитать коэффициенты активности отдельного вида ионов, но экспериментально можно определять лишь средние ионные коэффициенты активности. Поэтому для опытной проверки уравнения нужно перейти к средним ионным коэффициентам активности. Исходя из их определения (уравнение (15.24)), получим
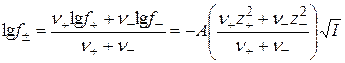 . (15.63)
. (15.63)
Учитывая, что система в целом электронейтральна, т.е. |n+z+| = |n–z–|, выражение в скобках представим в форме
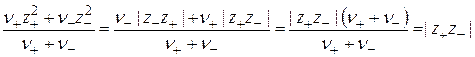 (15.64)
(15.64)
и окончательно получаем
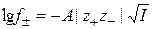 . (15.65)
. (15.65)
Это уравнение совпадает с эмпирически установленной закономерностью и является теоретическим обоснованием правила ионной силы Льюиса – Рендалла. Экспериментальная проверка уравнения (15.65) показала, что оно справедливо только для разбавленных растворов электролитов (I £ 0,01 – 0,02), при больших концентрациях наблюдаются значительные расхождения экспериментально определяемых величин с теоретически рассчитанными.
Ограниченность теории Дебая – Гюккеля предопределена, прежде всего, недостатками самой модели, использованной при создании теории.
Согласно модели, рассматривается только электростатическое межионное взаимодействие, которое вместе с тепловым движением приводит к образованию сферической ионной атмосферы. Это допущение справедливо только для разбавленных растворов. Увеличение концентрации приводит к уменьшению межионного расстояния, начинают проявляться силы межмолекулярного взаимодействия, например, силы Ван-дер-Ваальса, а размер ионной атмосферы становится соизмеримым с размерами самого иона. В этом случае уже нельзя рассматривать ионы как точечные заряды.
При расчетах используется разложение показательной функции в ряд с ограничением двумя или одним членом разложения, что соответствует ограничению разбавленными растворами.
Допущение о равенстве диэлектрической проницаемости раствора диэлектрической проницаемости растворителя также справедливо только для разбавленных растворов. Наконец, в теории не учитывается взаимодействие ионов с растворителем.
Все это приводит к тому, что теория оказывается применимой к очень разбавленным растворам и уравнение (15.65) является первым приближением, или предельным законом теории Дебая – Гюккеля. Попытки усовершенствования теории предпринимались как Дебаем и Гюккелем, так и другими исследователями. Во втором приближении авторы теории учли, что ионы имеют конечные размеры, и расстояние между ними не может быть меньше некоторой минимальной величины. Полученное во втором приближении выражение для коэффициентов активности имеет вид:
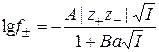 , (15.66)
, (15.66)
где 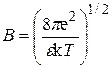 , a – расстояние наибольшего сближения ионов. Эту величину можно рассматривать как средний эффективный диаметр ионов. Значение a теоретически не вычисляется, а подбирается эмпирически.
, a – расстояние наибольшего сближения ионов. Эту величину можно рассматривать как средний эффективный диаметр ионов. Значение a теоретически не вычисляется, а подбирается эмпирически.
На практике часто используют также полуэмпирическое уравнение
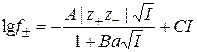 , (15.67)
, (15.67)
где C – эмпирическая постоянная. Это уравнение описывает зависимость коэффициентов активности от ионной силы в довольно широкой области концентраций.
15.6. Слабые электролиты.
Термодинамическая константа диссоциации
Как указывалось в разделе 15.1, существуют слабые электролиты, которые диссоциируют в растворе на ионы лишь частично, а их константа диссоциации определяется законом разведения Оствальда. Рассчитанная по уравнениям (15.1) – (15.3) константа сохраняет постоянное значение лишь в очень разбавленных растворах и изменяется при увеличении концентрации. Это связано с тем, что в теории Аррениуса не учитываются межионные взаимодействия, а в уравнениях используются концентрации вместо активностей.
Рассмотрим простейший пример диссоциации слабой кислоты НА в водном растворе:
НА + Н2О L Н3О+ + А–
Применяя к этому равновесию закон действия масс, получим для константы равновесия
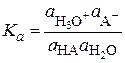 , (15.68)
, (15.68)
где ai – равновесные активности соответствующих частиц.
В разбавленных растворах активность воды можно считать постоянной и вынести ее в левую часть. Тогда
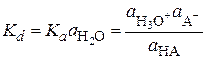 . (15.69)
. (15.69)
Величина Kd называется термодинамической константой диссоциации. В отличие от концентрационной константы диссоциации Kc, вычисленной по уравнению Оствальда, она не зависит от концентрации.
Значения термодинамической константы диссоциации можно определить по величинам концентрационных констант. Для рассматриваемого примера
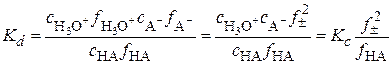 , (15.70)
, (15.70)
где ci – равновесные концентрации ионов и молекул, f± – средний ионный коэффициент активности, fHA – коэффициент активности молекул. При небольших ионных силах можно считать fHA » 1, тогда
lgKd = lgKc +2lgf±. (15.71)
Для разбавленных растворов f± можно выразить по уравнению Дебая – Гюккеля lgf± = – A , в котором для ионной силы взять величину 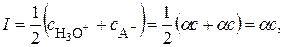 где c – общая концентрация кислоты. В этом случае после подстановки в уравнение (15.71) получим
где c – общая концентрация кислоты. В этом случае после подстановки в уравнение (15.71) получим
lgKd = lg 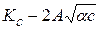 (15.72)
(15.72)
которое удобнее представить в форме
lgKc = lg 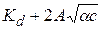 (15.73)
(15.73)
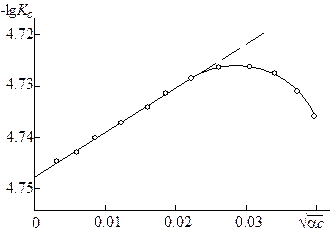 Рис. 15.5. Определение термодинамической константы диссоциации
Рис. 15.5. Определение термодинамической константы диссоциации
|
Это уравнение прямой в координатах lgKc – 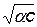 . Линейность соблюдается в области, где справедливо уравнение Дебая – Гюккеля, поэтому в области больших ионных сил наблюдаются отклонения от прямой (рис. 15.5). Экстраполяцией линейного участка на нулевую ионную силу получают значение термодинамической константы диссоциации. Необходимые для расчетов степени диссоциации определяются из экспериментальных данных, например, из величин молярной электропроводности (см. гл. 19). Обычно при этом получают приближенные степени диссоциации, а, следовательно, и констант диссоциации. Для точного определения используют метод последовательных приближений.
. Линейность соблюдается в области, где справедливо уравнение Дебая – Гюккеля, поэтому в области больших ионных сил наблюдаются отклонения от прямой (рис. 15.5). Экстраполяцией линейного участка на нулевую ионную силу получают значение термодинамической константы диссоциации. Необходимые для расчетов степени диссоциации определяются из экспериментальных данных, например, из величин молярной электропроводности (см. гл. 19). Обычно при этом получают приближенные степени диссоциации, а, следовательно, и констант диссоциации. Для точного определения используют метод последовательных приближений.
15.7. Единая количественная теория диссоциации электролитов
Деление электролитов на сильные и слабые сложилось в результате исследований водных растворов. Расширение круга исследованных растворителей и электролитов показало условность такого деления. Возникновение электролитного раствора является результатом целого ряда процессов, протекающих при растворении вещества. В наиболее общем виде схема равновесий, учитывающая взаимодействия и энергетику процессов, разработана Н. А. Измайловым.
Н. А. Измайлов исходил из того, что понятия о сильных и слабых электролитах относятся не к самим веществам, а к их состоянию в растворе, и все электролиты можно рассматривать с единой точки зрения.
Общая схема равновесий в растворе представляется в следующем виде:
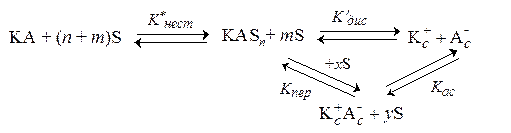
Молекулы электролита КА при попадании в растворитель S образуют вначале продукт присоединения КАSn, который подвергается дальнейшим превращениям: диссоциации с образованием сольватированных катиона Кc+ и аниона Аc– или ионизации с образованием ионных ассоциатов Кc+Аc– (нижний индекс "c" указывает, что частица сольватирована). Между ионами и ионными ассоциатами также устанавливается равновесие. Константы равновесия каждого процесса выражаются через активности соответствующих частиц, участвующих в этих равновесиях. В дальнейшем для упрощения записи индекс "c" опускаем.
Присоединение молекул растворителя к молекулам растворяемого вещества характеризуется константой нестойкости продукта присоединения

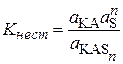 . (15.74)
. (15.74)
В разбавленных растворах активность растворителя можно считать постоянной, в этом случае
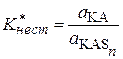 . (15.75)
. (15.75)
Равновесие между продуктом присоединения и сольватированными ионами определяется константой диссоциации K¢дис:
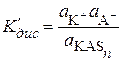 , (15.76)
, (15.76)
а равновесие между сольватированными ионами и ионными ассоциатами (ионными парами) – константой ассоциации
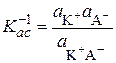 . (15.77)
. (15.77)
Константа Kпр является константой реакции превращения продукта присоединения в ионную пару:
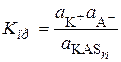 . (15.78)
. (15.78)
Величины 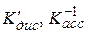 и Kпр связаны между собою:
и Kпр связаны между собою:
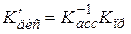 . (15.79)
. (15.79)
Обычные экспериментальные методы дают возможность определять активность ионов, а вычисленная по этим данным константа диссоциации является общей (обычной):
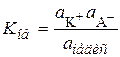 , (15.80)
, (15.80)
где aнедис представляет сумму концентраций свободных молекул электролита, молекул продуктов присоединения и ионных пар, умноженную на средний коэффициент активности:
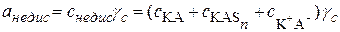 . (15.81)
. (15.81)
В разбавленных растворах коэффициенты активности всех частиц близки к единице, тогда
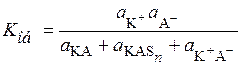 . (15.82)
. (15.82)
Выразим активности недиссоциированных частиц через активности ионов, используя уравнения (15.74) – (15.77):
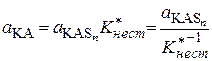 ;
; 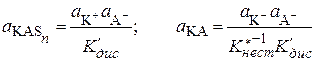 ;
;
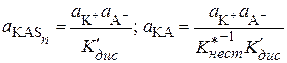 ;
; 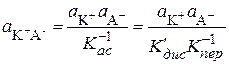 . (15.83)
. (15.83)
Подставив эти значения в уравнение (15.82), получим выражение для общей константы диссоциации:
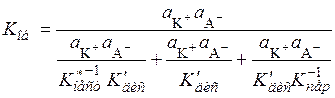 , (15.84)
, (15.84)
что дает окончательно
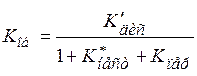 . (15.85)
. (15.85)
Из этого уравнения видно, что общая константа диссоциации выражена через постоянные величины и, следовательно, также является постоянной.
На основании уравнения (15.89) можно оценить влияние природы растворителя и растворенного вещества на силу электролита, рассмотрев некоторые частные случаи.
1) Если растворитель имеет высокую диэлектрическую проницаемость, то ассоциации ионов не происходит, Kасс ® 0, Kпр << 1 и
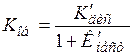 . (15.86)
. (15.86)
2) Если электролит полностью вступил во взаимодействие с растворителем, образовав продукт присоединения КАSn , то Kнест << 1 и
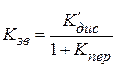 . (15.87)
. (15.87)
3) В растворителях с большим сродством к растворяемому веществу и высокой диэлектрической проницаемостью выполняются оба предыдущих условия, тогда
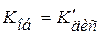 . (15.88)
. (15.88)
4) Если растворитель обладает большим сродством к ионам и высокой диэлектрической проницаемостью, то K¢дис ® ¥, Kасс ® 0 и вещество становится сильным электролитом:
Kоб ® ¥. (15.89)
Количественный учет влияния растворителя на силу электролита основан на рассмотрении работы кругового процесса, состоящего из переноса вещества КА из растворителя S в вакуум, диссоциации КА в вакууме на ионы и их переноса в раствор:
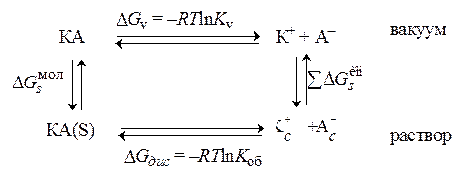
Изменение изобарного потенциала 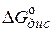 при диссоциации вещества в растворе в стандартных условиях связано с общей константой диссоциации
при диссоциации вещества в растворе в стандартных условиях связано с общей константой диссоциации 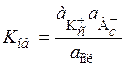 , где под молекулами понимаются все недиссоциированные частицы (КАSn , КА, Кс+Ас–).
, где под молекулами понимаются все недиссоциированные частицы (КАSn , КА, Кс+Ас–).
Диссоциация в вакууме характеризуется константой 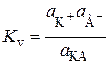 . Изменения изобарного потенциала при переносе ионов или молекул в раствор представляют собою энергии Гиббса сольватации ионов
. Изменения изобарного потенциала при переносе ионов или молекул в раствор представляют собою энергии Гиббса сольватации ионов 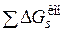 и молекул
и молекул 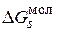 . Суммируя все изменения изобарного потенциала, приравнивая сумму нулю и потенцируя, получаем:
. Суммируя все изменения изобарного потенциала, приравнивая сумму нулю и потенцируя, получаем:
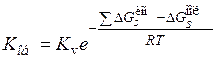 (15.90)
(15.90)
Изменения констант диссоциации при переходе от одного растворителя к другому при постоянной температуре определяются только разностью изменений энергий сольватации ионов и молекул:
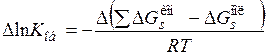 . (15.91)
. (15.91)
Приведенные зависимости являются общими и приложимы к процессам электролитической диссоциации солей, кислот и оснований.
| <== предыдущая лекция | | | следующая лекция ==> |
| Сглаживающие фильтры. | | | Генераторы синусоидального и несинусоидального сигналов. |
Дата добавления: 2015-12-16; просмотров: 2554;