Наследственно-дегенеративные заболевания ствола, мозжечка и спинного мозга
Наследственно-дегенеративные заболевания ствола, мозжечка и спинного мозга характеризуются медленно прогрессирующим течением с распадом функций, которые регулируются этими мозговыми структурами. Дебют заболеваний - в детском и юношеском возрасте.
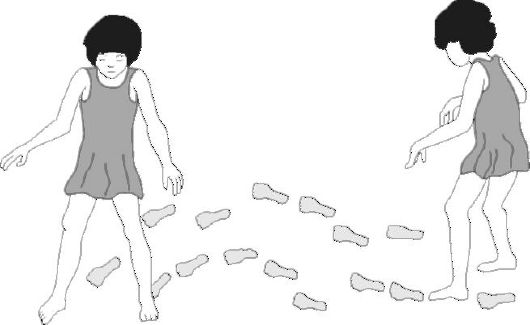
Рис. 5.Атактическая походка

Рис. 6.Атаксия в положении стоя

Рис. 7.Больная с Атаксией Фридрейха
Этиологияв большинстве случаев наследственная, заболевания передаются по аутосомнодоминантному или аутосомнорецессивному типу.
Патогенез.Прогрессирующее течение обусловлено атрофией нервной ткани в пределах пораженной области.
Классификация этих расстройств основана на генетических, клинических и патоморфологических данных.
Атаксия Фридрейха (G 11.1)
Атаксия Фридрейха (АФ) описана Н. Фридрейхом в 1863 г. Это наследственное заболевание, характеризующееся медленно прогрессирующей атаксией вследствие склеротического перерождения задних и боковых столбов спинного мозга, гипоплазии мозжечка и спинного мозга (рис. 5-7). Для него характерны атаксия, нистагм, кифосколиоз, деформация стопы. Больные отличаются особым дизморфическим статусом, имеют множество скелетных аномалий, часть из которых сформирована с рождения. Прибли- зительно у трех из четырех пациентов имеются высокий свод стопы (полая стопа), пальцы в виде барабанных палочек, атрофированы мелкие мышцы стопы. Кифосколиоз наблюдается в 75-90% случаев. Распространенность в популяции вариабельна - максимально до 10 случаев на 100 000 с высокой частотой гетерозиготного носительства мутантного гена - 1 на 120 человек.
Генетика.Заболевание передается аутосомно-рецессивным путем; ген картирован на хромосоме 9q13. Он кодирует митохондриальный белок фратаксин, расположенный на внутренней поверхности мембраны митохондрий и участвующий в обмене железа. В интроне патологического гена увеличена последовательность повторов ГАА (гуанинаденин-аденин). Количество ГАА-повторов находится в диапазоне от 6 до 29 у здоровых людей и от 120 до 1700 - у больных, причем размер повторов коррелирует с возрастом дебюта и тяжестью болезни. Патологически удлиненный аллель генетически нестабилен и способен к дальнейшей экспансии при его передаче в следующее поколение. В результате мутации снижается уровень нормального фратаксина, железо откладывается внутри митохондрий, происходит необратимое повреждение функции митохондрий и нарушение окислительного фосфорилирования. В результате гибнут клетки энергозависимых мишеней (мозга, сердца, поджелудочной железы, почек, печени). Таким образом, атаксия Фридрейха - это митохондриальное заболевание, связанное с мутацией ядерного генома. У гетерозигот неврологических симптомов не наблюдается.
Патогенезсвязан с дегенерацией длинных проводников спинного мозга. Наряду с периферическими нервами также могут поражаться продолговатый мозг и, реже, мозжечок. В этих областях выявляются аксональная дегенерация, демиелинизация и компенсаторный глиоз. Дегенеративные изменения наиболее выражены в столбах Кларка и зубчатых ядрах мозжечка, но поражаются также ядра продолговатого мозга и клетки Пуркинье. Апоптоз нейронов и глиоз отмечаются в вестибулярных и слуховых ядрах. В миелиновой оболочке проводников снижен уровень протеолипидов. Возможна патология со стороны внутренних органов: кардиомегалия с гипертрофией миоцитов, а в поджелудочной железе - хронический интерстициальный фиброз и воспалительная инфильтрация. Нередко выявляется сахарный диабет.
Патоморфология.Выявляется гибель клеток столбов Кларка и начинающихся от них спиноцеребеллярных трактов, а также (в поздней стадии болезни) дегенерация ядер III, V, IX-X, XII пар черепных нервов, клеток Пуркинье, зубчатого ядра и верхней ножки мозжечка. В указанных областях выявляются аксональная дегенерация, демиелинизация и компенсаторный глиоз.
Клинические проявления.Возраст дебюта вариабелен, однако в одной семье заболевание начинается в одном возрасте. Первые симптомы могут отмечаться уже в 2-летнем возрасте, средний возраст дебюта - 10 лет. Течение характеризуется появлением новых симптомов, относительно быстрым прогрессированием процесса и сочетанием типичных неврологических и экстраневральных нарушений.Дети начинают ходить после года, часто падают. При более позднем дебюте возникает пошатывание, нарушена ходьба в темноте (признак заднестолбовой атаксии). Вскоре к атаксии при ходьбе присоединяются дискоординация рук, изменение почерка, слабость в ногах. Со стороны черепных нервов обнаруживаются нарушения остроты зрения из-за атрофии зрительных нервов, нистагм (в 20-40% случаев), а также снижение слуха. Кроме того, могут наблюдаться подергивания глазных яблок (миоклонии). Атрофия зрительных нервов может быть врожденной или быстро нарастает на первом году жизни. У 40% больных нарушено восприятие цветов. Вестибулярные расстройства возникают рано, на поздних этапах болезни встречаются приблизительно у 50% пациентов. Также типична глухота, вызванная дегенерацией слуховых нейронов. Наиболее примечательным симптомом является комбинированная мозжечковосенситивная атаксия, вызванная поражением мозжечка и задних столбов с их чувствительными проводниками. Она более выражена в ногах, чем в руках, и выявляется при исследовании походки и статики ребенка. Можно выявить отсутствие вибрационной и проприоцетивной чувствительности, в далеко зашедших случаях в дистальных отделах конечностей нарушены другие виды чувствительности. При неврологическом обследовании выявляется арефлексия коленных и ахилловых рефлексов. Возникают слабость дистальных мышц нижних конечностей и атрофия мелких мышц рук и ног. Часты жалобы на боли, судороги и парестезии в конечностях. В развернутой клинической стадии нарушения координации нарастают, к ним присоединяется слабость и атрофия мышц ног, а затем и рук, вплоть до тетрапареза. Речь становится раскатистой в результате несогласованности дыхания и фонации. По поводу деменции мнения противоречивы: для детей умственная отсталость и деменция нехарактерны.
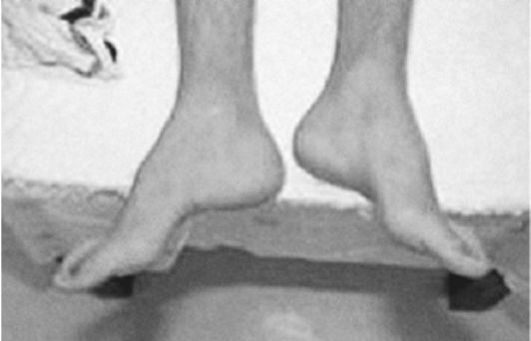
Рис. 8.Деформация стоп по типу Фридрейха
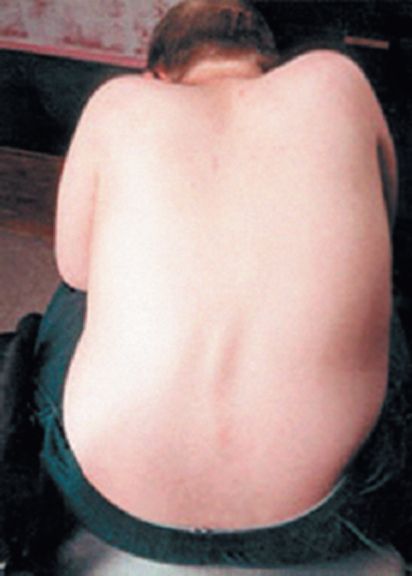
Рис. 9.Сколиоз при атаксии Фридрейха
Расстройства функций тазовых органов характерны для финальной стадии болезни, а ранним симптомом могут быть внезапные позывы к мочеиспусканию. Среди экстраневральных проявлений болезни Фридрейха необходимо выделить поражение сердца, которое встречается более чем у 90% больных. Характерна прогрессирующая гипертрофическая или дилатационная кардиомиопатия. Она проявляется болями в области сердца, сердцебиением, одышкой при физической нагрузке, систолическим шумом и другими симптомами. Более чем у половины больных кардиомиопатия является непосредственной причиной смерти. Деформации стоп - «стопа Фридрейха» - не патогномонична для болезни Фридрейха (рис.8) и встречается при некоторых других дегенеративных заболеваниях нервной системы, например при невральной амиотрофии Шарко-Мари, спастической параплегии Штрюмпеля и др. Нередок также сколиоз (рис.9). К экстраневральным проявлениям болезни Фридрейха относятся эндокринные расстройства (сахарный диабет, гипогонадизм, инфантилизм, дисфункция яичников).
Неврологические симптомы прогрессируют медленно, с продолжительностью заболевания до 20 лет, хотя возможно более быстрое течение болезни. Иногда наблюдаются периоды стабилизации состояния. Сопутствующие инфекции ухудшают течение заболевания и способствуют появлению новых симптомов. Больной с далеко зашедшей болезнью прикован к постели, страдает дисфагией и другими бульбарными симптомами. Смерть наступает от истощения или, чаще, от миокардита с тяжелой сердечной недостаточностью. При хорошем уходе пациенты могут доживать до 40-50 лет.
Дополнительные методы исследования. При исследовании зрительных вызванных потенциалов выявляются генерализованное снижение амплитуды потенциалов и удлинение времени их появления. Уменьшение амплитуды, вероятно, является последствием распада волокон зрительных путей. Соматосенсорные вызванные потенциалы, регистрируемые от надключичных отведений, отличаются от нормальных уже на самых ранних стадиях болезни, но они не сопровождаются снижением проводимости по периферическому нерву.
МРТ может выявить расширение IV желудочка и атрофию верхнего червя, ствола и спинного мозга. При проведении ЭКГ и Эхо-КГ признаки миокардита выявляются в 80-90% случаев. Особенно часто отмечаются нарушения проводимости, вплоть до полной блокады, и гипертрофия межжелудочковой перегородки. При цитохимическом исследовании ферментов-дегидрогеназ лимфоцитов выявляется достоверное снижение сукцинатдегидрогеназы (СДГ), α-глицерофосфадегидрогеназы (ГФДГ), глутаматдегидрогеназы (ГДГ), лактатдегидрогеназы (ЛДГ), малатдегидрогеназы (МДГ) и др. Необходимо иметь в виду, что при молекулярно-генетическом обследовании пациентов с клинически типичными проявлениями не у всех обнаруживается увеличение тринуклеотида ГАА, расширение аллеля. Возможна точечная мутация или делеция в гене на обеих хромосомах. Описана аутосомно-рецессивная форма мозжечковой атаксии, при которой сухожильные рефлексы сохранены и нет атрофии зрительных нервов, диабета и нарушений со стороны сердца. Симптомы появляются в возрасте от 18 мес до 20 лет, течение медленнее, чем при классической форме. Почти у половины пациентов с такой клинической картиной можно найти увеличение повторов ГАА.
Диагноз.В типичном случае клинический диагноз ставится на основании имеющихся с раннего детства прогрессирующей атаксии, скелетных деформаций, нарушений зрительных вызванных потенциалов и кардиопатии. Диагноз подтверждается генетически (определение размера повторов ГАА).
Дифференциальный диагнозв первую очередь должен проводиться со второй по частоте встречаемости прогрессирующей атаксией с началом в детском возрасте - атаксией-телеангиоэктазией (болезнью Луи-Бар). Клинически она отличается наличием на коже телеангиоэктазий (чрезмерного локального расширения мелких сосудов, преимущественно прекапилляров и капилляров), отсутствием скелетных аномалий, частыми и тяжело протекающими инфекциями дыхательных путей, отсутствием или крайне низким уровнем IgA, высоким уровнем альфа-фетопротеина. На МРТ выявляется гипоплазия мозжечка, чаще его червя.
Лечениеатаксии Фридрейха не разработано. Применяют препараты, поддерживающие функцию митохондрий (табл. 10). Рекомендуется одновременное назначение препаратов, повышающих активность дыхательной цепи митохондрий, кофакторов энзимных реакций энергетического обмена, антиоксидантов. Пациенты чувствуют себя лучше при ограничении количества углеводов в пище до 10 г/кг, поскольку они являются своеобразной «провокацией», усиливающей дефект энергетического обмена. Дети с АФ могут оставаться активными максимально долго, занимаясь лечебной физкультурой, выполняя комплексы корректирующих упражнений, направленных на укрепление силы мышц и нормализацию баланса. При такой программе упражнений кардиомиопатия не развивается. Ортопедическое хирургическое лечение скелетных деформаций, особенно прогрессирующего сколиоза, показано, если неэффективен ортопедический корсет.
Таблица 10.Медикаментозные препараты, применяемые для лечения БФ

Прогноз.Болезнь Фридрейха характеризуется неуклонно про- грессирующим течением, длительность болезни может варьировать в широких пределах, но чаще не превышает 20 лет. Непосредственными причинами смерти могут быть сердечная и легочная недостаточность, инфекционные осложнения.
Спиноцеребеллярные атаксии [оливопонтоцеребеллярная дегенерация]. Оливопонтоцеребеллярная дегенерация - генетически и клинически гетерогенные состояния. Для них характерны прогрессирующая мозжечковая атаксия, тремор, головокружение, дизартрия, снижение глубокой чувствительности, глазодвигательные нарушения и пирамидные симптомы. Реже наблюдаются гиперкинезы, симптомы периферического паралича и тазовые нарушения. Патологический процесс поражает нейроны коры мозжечка, ядра варолиева моста и нижних олив, а также в той или иной степени спинной мозг и базальные ядра. Степень тяжести обусловлена характером мутации и длиной патологического гена. В результате молекулярно-генетических исследований в настоящее время выделено более 10 типов атаксий, которые получили название спиноцеребеллярных атрофий (СЦА). Но даже при молекулярно-генетическом исследовании приблизительно у половины семей с аутосомно-доминантной мозжечковой атаксией не обнаруживается ни одна из известных мутаций. Тем не менее диагноз аутосомнодоминантной мозжечковой атаксии базируется на идентификации генетической мутации. Средний возраст дебюта этих заболеваний приходится на четвертое десятилетие жизни, однако ряд состояний встречается у детей.
Этиология.Гены картированы на хромосомах: СЦА1 - на 6р22-23, СЦА2 - на 12q24.1, СЦАЗ - на 14q32.1, СЦА4 - на 16q21, СЦА5 - на 11q13, СЦА7 - на 3р12-13, СЦА8 - на 13q 21, СЦА10 - на 22q13. Ген формы СЦА6 картирован на хромосоме 19р13. И только при этой форме установлен механизм работы гена, который кодирует альфа-1-субъединицу вольтаж-зависимых кальциевых каналов. Механизм мутаций при СЦА заключается в патологическом увеличении числа тринуклеотидных повторов. Длина повторов нарастает из поколения в поколение, поэтому чем длиннее повтор, тем раньше дебютирует заболевание и тем тяжелее оно протекает (антиципация). Такой характер повреждения гена и проявления болезни характерен для болезни Гентингтона, миотонической дистрофии, спинальнобульбарной амиотрофии Кеннеди и многих других неврологических заболеваний. Распространенность отдельных генетических форм СЦА варьирует в различных популяциях. В Северной Америке преобладающей формой является СЦА3, в России чаще всего встречается СЦА1. При этой форме увеличенная полиглутаминовая последовательность провоцирует нейрональную дегенерацию. Обычно клиническая картина дебютирует в возрасте до 15 лет, причем у мальчиков раньше, так как повторы в большей степени удлиняются при наследовании по отцовской линии. Характерны атаксия, офтальмоплегия, пирамидные и экстрапирамидные симптомы. Морфологически выявляется атрофия мозжечка и его ножек, а также основания моста. Наиболее сильно страдают клетки Пуркинье и нейроны зубчатого ядра, а также базальные ядра, спинной мозг, сетчатка глаза и периферическая нервная система.
Диагноз и дифференциальный диагнозосновывается на времени дебюта, характерном сочетании симптомов и скорости их развития у детей, чьи родители страдают прогрессирующей атаксией.
Семейная спастическая параплегия. Заболевание передается аутосомно-доминантным, аутосомно-рецессивным или Х-сцепленным путем.
Патогенез.Основные изменения происходят в спинном мозге. Аксональная дегенерация пирамидальных путей всегда максимально выражена в дистальных отделах. В пораженных проводниках разрушаются осевой цилиндр и миелиновая оболочка. Поражаются также восходящие пути, в особенности задние столбы, спиноцеребеллярные волокна и клетки спинномозговых узлов, которые дегенерируют на фоне пролиферации глии. Признаков первичной демиелинизации не обнаруживается. При биопсии мышц можно обнаружить рваные красные волокна.
Клиническая картинапри всех формах сходна. При рецессивных вариантах болезни средний возраст развития полной клинической картины - 11,5 года, а при доминантных - 20 лет. Однако у 40% пациентов первые симптомы появляются до 5-летнего возраста. Дети начинают позднее ходить, выявляются шаткость и неуклюжесть, перекрещивание ног в виде ножниц. Мышечный тонус в ногах и сухо- жильные рефлексы повышены, выявляются патологические стопные симптомы. Это заболевание часто протекает под маской детского церебрального паралича. Следует заметить, что при ССП не наблюдается атрофии мышц и, несмотря на поражение задних столбов, вибрационная чувствительность не нарушена.Как правило, течение болезни очень медленное, причем быстрее прогрессирует рецессивная форма. Если ребенок страдает той или иной доминантной формой, его состояние относительно стабильно до 30 лет. Верхние конечности часто остаются интактными вплоть до терминальной стадии. Соматические нарушения на ранних стадиях болезни не наблюдаются. В некоторых семейных случаях спастическая параплегия сочетается с деменцией, судорогами, гиперкинезами, невритом зрительного нерва, патологией сердца, гипопигментацией кожи.
Диагноз.При отсутствии семейного анамнеза диагноз наследствен- ной параплегии ставится методом исключения. Время проведения по двигательным и чувствительным нервам не нарушено, соматосенсорные вызванные потенциалы снижены, причем не только у больных, но и у клинически здоровых членов семьи. Прогрессирующее развитие симптомов опровергает диагноз ДЦП. Чувствительные расстройства и нарушение функций сфинктеров, обычно характерные для опухоли спинного мозга, редко встречаются на ранних стадиях болезни. Однако при отсутствии убедительного семейного анамнеза требуется проведение МРТ для исключения новообразований спинного мозга.
Лечение.Ввиду медленного прогрессирования болезни должна применяться активная программа физиотерапии и лечебной физкультуры для предотвращения контрактур.
Петрухин А.С. Детская неврология ,2012г
6. СОСТАВЛЕНИЕ РОДОСЛОВНОЙ
Составление родословной начинается со сбора сведений о семье, и прежде всего со сбора сведений о пробанде— индивиде, который является предметом интереса исследователя (врача, педагога). Чаще всего это больной или носитель изучаемого признака. Однако за медико-генетической консультацией могут обращаться и здоровые индивиды. В этом случае используется термин «консультирующийся». В графическом изображении родословной пробанд отмечается соответствующим знаком и стрелкой, которая идет снизу вверх и слева направо. Дети одной родительской пары (братья и сестры) называются сибсами.Если сибсы имеют только одного общего родителя, они называются полусибсами.Различают единоутробных (общая мать) и единокровных (общий отец) сибсов. Семьей в узком смысле называют родительскую пару и их детей (ядерная семья), но иногда и более широкий круг кровных родственников. В последнем случае лучше использовать термин «род».
Обычно родословная собирается в связи с изучением одного или нескольких заболеваний (признаков). Врач или генетик всегда интересуется каким-то конкретным заболеванием или признаком.
В зависимости от цели исследования родословная может быть полной или ограниченной: она может отражать либо клинические признаки, либо генетический статус членов родословной. В любом случае нужно стремиться к наиболее полному составлению родословной по восходящему, нисходящему и боковым направлениям. Чем больше поколении вовлекается в родословную, тем больше информации она может содержать. Однако ее обширность может обусловить появление в ней ошибочных данных. Для уточнения сведений привлекаются различного рода медицинская документация, фотографии родственников, результаты дополнительных исследований. Чем больше глубина и широта генеалогического поиска, тем ценнее и надежнее получаемая информация.
Для наглядности собранные данные изображают в виде определенных символов, некоторые из которых представлены на рис. IХ.1.

Рис. 1Х.1. Символы, наиболее часто используемые при составлении родословных:
1 - лицо мужского пола; 2 - лицо женского пола; 3 - больные; 4 - брак; 5 - кровнородственный брак; 6 - сибсы; 7 - единоутробные сибсы; 8 - единокровные сибсы; 9 - монозигошые близнецы; 10 - дизиготные близнецы; 11 - усыновление; 12 - пол неизвестен; 13 - выкидыш; 14 - медицинский аборт; 15 - умершие; 16 - пробанд; 17 - гетерозиготные индивиды; 18 - гетерозиготная носительница рецессивного гена в Х-хромосоме; 19 - беременность; 20 - бесплодный брак; 21 - лично обследован
Под «клинической» родословной понимают отображение наследования конкретного заболевания или нескольких заболеваний. Максимальное число заболеваний (признаков) в одном символе, т.е. у одного индивида, в графическом изображении не должно превышать четырех нозологических форм или признаков. Если клиническая родословная посвящена анализу только одного конкретного заболевания, то обозначения 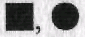 соответствуют изображению больного мужского пола и больной женского пола. Если в клинической родословной прослеживаются два заболевания, например гипертоническая болезнь и ожирение, то обычно используют следующие обозначения: каждый символ делят на две равные части, при этом больные первым заболеванием (гипертонической болезнью) обозначаются
соответствуют изображению больного мужского пола и больной женского пола. Если в клинической родословной прослеживаются два заболевания, например гипертоническая болезнь и ожирение, то обычно используют следующие обозначения: каждый символ делят на две равные части, при этом больные первым заболеванием (гипертонической болезнью) обозначаются 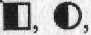 а больные с ожирением как
а больные с ожирением как 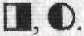 В данной родословной символ
В данной родословной символ  обозначал бы индивида мужского пола, страдающего и гипертонической болезнью, и ожирением одновременно.
обозначал бы индивида мужского пола, страдающего и гипертонической болезнью, и ожирением одновременно.
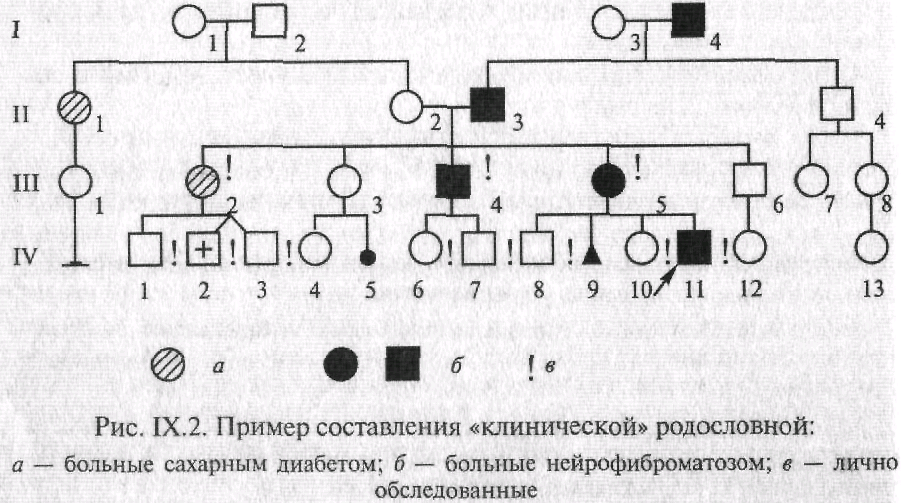
В некоторых случаях для отображения в родословной различных заболеваний используют различающиеся виды штриховки элементов (рис. IX.2). Графическое изображение родословной дополняется обязательными разделами: «Условные обозначения» и «Легенда родословной».
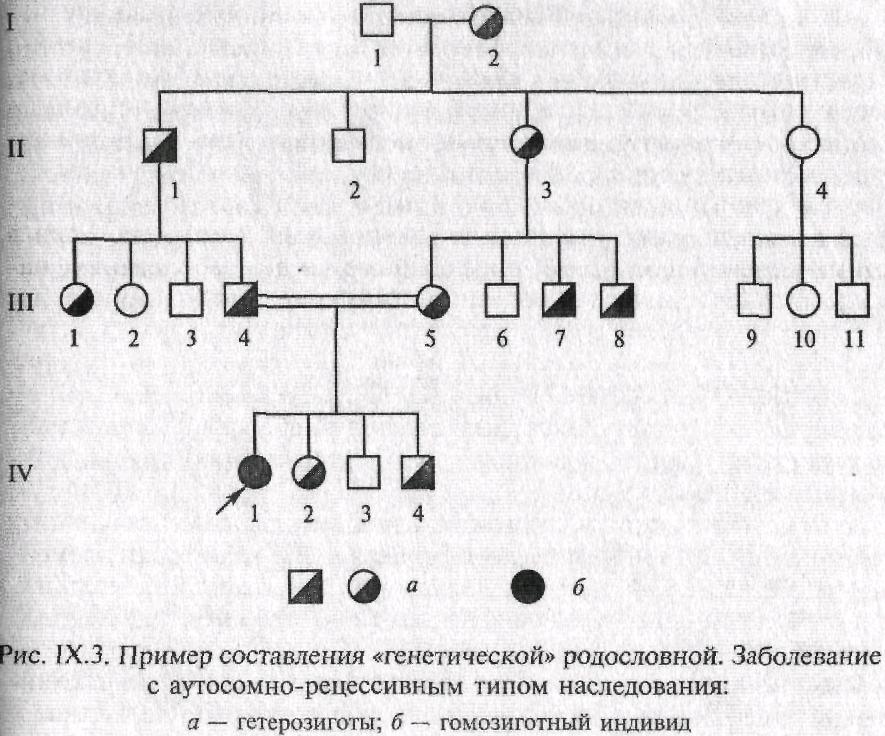
Условныеобозначения — это перечень символов, использованных при графическом представлении родословной. Как правило, применяют стандартные значки-символы (рис. IХ.1). Однако в зависимости от задач, целей и особенностей родословных составитель вправе использовать оригинальные (собственные) обозначения с обязательным их объяснением, чтобы исключить возможность неправильных толкований данных. Для пояснения принципов обозначения и составления родословных приведены два примера (рис. 1Х.2 и 1Х.З).
Легендародословной является обязательным элементом описания родословной. Она включает:
1) подробное описание каждого члена родословной, сведения о котором обязательны или существенны для понимания характера наследования заболевания (признака) или особенностей клинического проявления;
2) перечень источников медицинских и других сведений с содержательной информацией;
3) указание на характер патологического процесса или его локализацию (например, у некоторых членов родословной диагностирована изолированная злокачественная опухоль желудка, у других — множественные неоплазии);
4) указание на время начала заболевания и особенности течения;
5) указание на возраст и причину смерти;
6) описание методов диагностики и идентификации (например, качественный или количественный характер описываемого (признака).
Таким образом, «Легенда родословной» — это информация о членах родословной с подробным изложением любых, но обязательно существенных для анализа сведений.
Поколения обозначаются римскими цифрами сверху вниз, обычно они ставятся слева от родословной. Последнее поколите предков, по которому собрана информация, обозначается как I поколение. Арабскими цифрами нумеруются все элементы одного поколения (весь ряд) слева направо, последовательно. Братья и сестры располагаются в родословной в порядке рождения. Таким образом, каждый член родословной имеет свои координаты, например в родословной, представленной на рис. IХ.2, дедушка пробанда по материнской линии — II-3, болен нейрофиброматозом.
Все индивиды одного поколения должны располагаться строго в один ряд. «Подвешивание» символов между рядами поколений является грубой ошибкой. Если родословная обширна, то поколения можно располагать не горизонтальными рядами, а концентрическими кругами (рис. IX.4). В родословной важно отмечать лично обследованных на присутствие признака заболевания или заболевания.
Исследователь должен стремиться к получению объективного первичного материала, который кладется в основу статистического и генетического анализа.
Дата добавления: 2015-10-05; просмотров: 3030;