Лекция № 4
Кислотно-основные свойства органических соединений, ионизация. Роль ионизации в проявлении биологической активности
Согласно теории электролитической диссоциации Аррениуса (1887 г.), кислотами являются вещества, диссоциирующие в водных растворах с образованием в качестве катионов только катионов водорода Н+, основаниями – вещества, при диссоциации которых в качестве анионов образуются только гидроксид-анионы ОН-. Эти определения справедливы для тех реакций, которые идут в водных растворах. В то же время было известно большое число реакций, приводящих к образованию солей, но реагирующие вещества не являлись кислотами и основаниями согласно теории Аррениуса. В 1923 г. были предложены две теории кислот и оснований: протолитическая теория Бренстеда и Лоури, а также электронная теория Льюиса.
Согласно протолитической теории, кислоты – это ионы или молекулы, способные отдавать катион водорода, т.е. вещества, являющиеся донорами протона. Основания – это молекулы или ионы, способные присоединять катион водорода, т. е. вещества, являющиеся акцепторами протона или донорами пары электронов, необходимой для присоединения протона. По этой теории кислота и основание составляют сопряженную пару и связаны уравнением: кислота ↔ основание + Н+.
В протолитической теории понятия кислот и оснований относятся лишь к функции, которую выполняет вещество в данной реакции. Одно и то же вещество, в зависимости от реакционного партнера, может выполнять функцию как кислоты, так и основания:
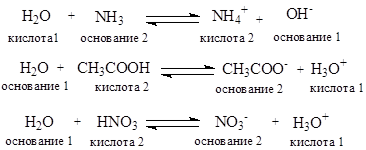
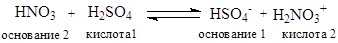
Обычно кислотность определяется по отношению к воде как основанию. Количественная оценка кислотности (силы кислоты) проводится сравнением констант равновесия реакций по переносу протона от кислоты к основанию.
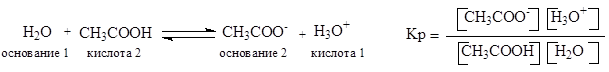
Концентрация воды практически не изменяется, поэтому, умножив правую и левую части этого равенства на [Н2О], получим следующее выражение:
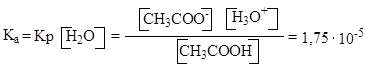
Ка – константа кислотности, чем больше значение константы кислотности, тем кислота сильнее. На практике для удобства часто используют не константу кислотности, а отрицательный десятичный логарифм константы кислотности, называемый показателем кислотности рКа = – lg Ка. Для уксусной кислоты константа кислотности Ка =1,75 ·10-5, а показатель кислотности рКа = 4,75. Чем меньше значение рКа, тем кислота сильнее. У более сильной муравьиной кислоты эти значения равны, соответственно: Ка =1,7 ·10-4, рКа = 3,77.
Сравнительный анализ силы кислот (качественная оценка) проводят путем сопоставления устойчивости соответствующих кислотам сопряженных оснований (анионов). Чем стабильнее сопряженный кислоте анион (основание), тем сильнее сопряженная ему кислота. Стабильность анионов зависит от степени делокализации отрицательного заряда – чем в большей степени делокализован отрицательный заряд, тем стабильнее анион, тем сильнее сопряженная кислота.
Степень делокализации отрицательного заряда зависит от следующих факторов:
1) от природы атома кислотного центра, т.е. от его электроотрицательности и радиуса (поляризуемости);
2) от характера связанного с ним радикала;
3) от электронного строения аниона;
4) от влияния растворителя.
Влияние природы атома кислотного центра
В зависимости от природы кислотного центра различают: ОН-кислоты (спирты, фенолы, карбоновые кислоты), SH-кислоты (тиолы), NH-кислоты (амиды, амины), СН-кислоты (углеводороды). Для рассмотрения влияния электроотрицательности атома кислотного центра возьмем соединения, в которых атомы кислотного центра связанны с одинаковыми заместителями: СН4, NH3, H2O. Все атомы кислотных центров расположены в одном периоде, от углерода к кислороду увеличивается электроотрицательность, в этом же направлении происходит увеличение полярности связей и уменьшение прочности связей атомов кислотных центров с атомом водорода. Таким образом, можно говорить о том, что при переходе от метана к воде увеличивается способность соединений отщеплять катион водорода, т.е. быть донорами протона. При этом в ряду возникающих анионов Н3С-, H2N-, HO- увеличивается их стабильность, так как с увеличением электроотрицательности атома кислотного центра увеличивается его способность удерживать отрицательный заряд. В ряду соединений метан – аммиак – вода происходит усиление кислотных свойств. Сравнивая с этими тремя молекулами молекулу Н2S, необходимо учитывать не только электроотрицательность атома серы, но и атомный радиус серы и поляризуемость этого атома. По электроотрицательности сера занимает промежуточное положение между углеродом и азотом. Исходя из приведенных выше рассуждений можно было бы ожидать, что кислотные свойства Н2S будут выражены сильнее, чем у метана, но слабее, чем у аммиака. Но атом серы среди рассматриваемых кислотных центров имеет самый большой атомный радиус (как элемент третьего периода), что обуславливает большую длину связи с атомом водорода и ее меньшую прочность. Кроме того, больший, чем у других кислотных центров, атомный радиус обеспечивает большую поляризуемость атома серы, т.е способность аниона HS- рассредотачивать электронную плотность и отрицательный заряд в большем объеме, что увеличивает стабильность этого аниона в сравнении с рассмотренными выше. Таким образом, эти кислоты и соответствующие им сопряженные основания (анионы) можно расположить в ряд по усилению кислотных свойств и увеличению стабильности анионов:
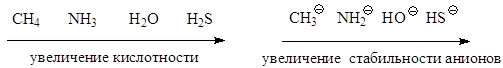
Аналогичная картина наблюдается и для соединений, в которых атом кислотного центра связан с одинаковым органическим радикалом:
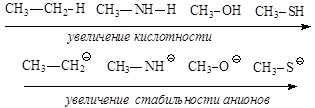
С-Н кислоты проявляют самые слабые кислотные свойства, хотя алканы, алкены и алкины несколько различаются по кислотности.
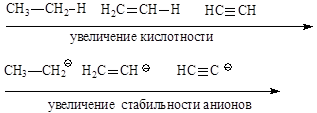
Увеличение кислотности в этом ряду обусловлено увеличением электроотрицательности атома углерода при переходе от sp3- к sp-гибридизации.
Влияние заместителей, связанных с кислотным центром
Электроноакцепторные заместители увеличивают кислотность соединений. Смещая на себя электронную плотность, они способствуют увеличению полярности и уменьшению прочности связи атома кислотного центра с атомом водорода, облегчают отщепление протона. Смещение электронной плотности к электроноакцепторному заместителю приводит к большей делокализации отрицательного заряда в анионе и увеличению его стабильности.
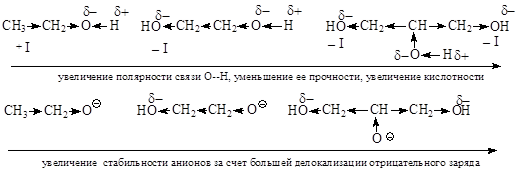
Электронодонорные заместители уменьшают кислотность соединений, так как смещают электронную плотность от себя, что приводит к локализации отрицательного заряда на атоме кислотного центра в анионе и уменьшению его устойчивости, увеличению его энергии, что затрудняет его образование.
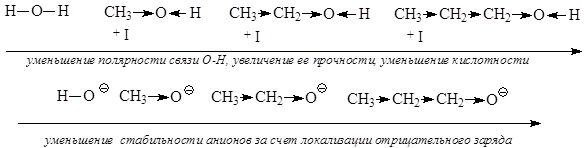
Влияние электронного строения анионов
На степень делокализации отрицательного заряда в анионе и его стабильность оказывает сильное влияние наличие сопряженной системы и проявление мезомерного эффекта. Делокализация отрицательного заряда по системе сопряжения приводит к стабилизации аниона, т.е к усилению кислотных свойств молекул.
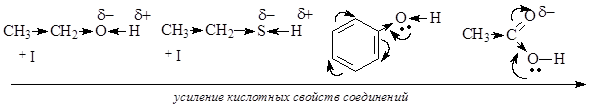
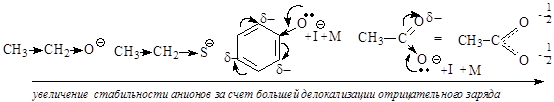
Молекулы карбоновых кислот и фенола образуют более стабильные анионы и проявляют более сильные кислотные свойства, чем алифатические спирты и тиолы, в которых не проявляется мезомерный эффект.
Влияние растворителя
На проявление кислотных свойств соединения влияние растворителя может быть значительным. Так, например, соляная кислота, являющаяся сильной кислотой в водном растворе, практически не проявляет кислотных свойств в бензольном растворе. Вода, как эффективный ионизирующий растворитель, сольватирует образующиеся ионы, тем самым стабилизирует их. Молекулы бензола, являясь неполярными, не могут вызвать значительной ионизации молекул хлороводорода и не могут стабилизировать за счет сольватации образующиеся ионы.
В протолитической теории кислот и оснований различают два типа оснований – p-основания и n-основания (ониевые основания).
p-Основания – это соединения, которые для образования связи с протоном предоставляют пару электронов p-связи. К ним относятся алкены, диены, ароматические соединения. Они являются очень слабыми основаниями, так как пара электронов не свободна, а образует p-связь, т.е принадлежит обоим атомам. Для образования s-связи с протоном сначала нужно разорвать p-связь, что требует затрат энергии.
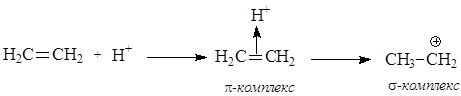
n-Основания (ониевые основания) – это молекулы или ионы, которые для образования связи с протоном предоставляют неподеленную пару р-электронов. По природе оснóвного центра различают: аммониевые основания, оксониевые основания и сульфониевые основания.
Аммониевые основания – это соединения, в которых центром основности является атом азота с неподеленной парой р-электронов (амины, амиды, нитрилы, азотсодержащие гетероциклы,имины и др.)
Оксониевые основания – это соединения, в которых центром основности является атом кислорода с неподеленной парой р-электронов (спирты, простые и сложные эфиры, альдегиды, кетоны, карбоновые кислоты и др.)
Сульфониевые основания – это соединения, в которых центром основности является атом серы с неподеленной парой р-электронов (тиоспирты, тиоэфиры и др.).
Cилу основания В в воде можно оценить, рассматривая равновесие:
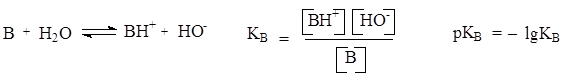
Константу основности КВ, так же как и константу кислотности Ка, для удобства выражают величиной рКВ, численно равной отрицательному десятичному логарифму константы основности. Чем больше константа основности КВ и чем меньше рКВ, тем сильнее основание.
Для количественной оценки силы оснований используют также показатель кислотности рКа сопряженной кислоты ВН+, обозначаемый рКВН+:
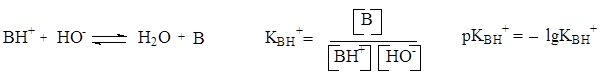
Чем меньше значение КВН+ и чем больше значение рКВН+, тем сильнее основание. Величины рКВ в воде можно переводить в рКВН+, используя соотношение: рКВ + рКВН+ = 14.
Сила оснований зависит от: 1) природы атома основного центра – электроотрицательности и поляризуемости (от радиуса атома); 2) от электронных эффектов заместителей, связанных с основным центром; 3) от влияния растворителя.
Влияние природы атома основного центра
С увеличением электроотрицательности атома основного центра сила оснований уменьшается, так как чем больше электроотрицательность, тем сильнее атом удерживает свою неподеленную пару электронов, и таким образом труднее ее предоставляет для образования связи с протоном. Исходя из этого, оксониевые основания слабее аммониевых, содержащих одинаковые заместители у основного центра:
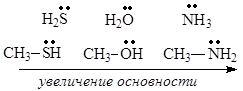
Сульфониевые основания, содержащие одинаковые заместители у основного центра, проявляют еще более слабые основные свойства. Атом серы, хотя и менее электроотрицателен, чем атомы кислорода и азота, имеет больший атомный радиус и характеризуется большей поляризуемостью, поэтому труднее предоставляет неподеленную пару электронов внешнего слоя для образования связи с протоном.
Влияние заместителей, связанных с основным центром
Электронодонорные заместители, смещая электронную плотность к атому основного центра, облегчают присоединение протона, тем самым усиливают основные свойства. Электроноакцепторные заместители, смещая на себя электронную плотность, уменьшают ее на основном центре, чем затрудняют присоединение протона и ослабляют основные свойства:
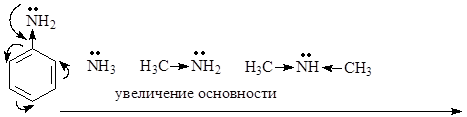
Влияние растворителя:
Поскольку увеличение силы основания связано с возрастанием способности присоединять протон и, следовательно, с увеличением на основном центре частичного отрицательного заряда, можно ожидать повышения основности в ряду аммониевых оснований NH3 < RNH2 < R2NH < R3N в результате усиления индуктивного эффекта при последовательном увеличении числа алкильных групп. В действительности, однако, ряд аминов имеет следующие значения рКВН+:
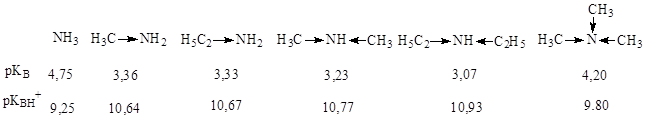
Как и следовало ожидать, введение алкильной группы в молекулу аммиака значительно повышает основность соединений, причем этильная группа оказывает несколько больший эффект, чем метильная. Введение второй алкильной группы приводит к дальнейшему повышению основности, однако эффект от ее введения выражен значительно слабее. Введение же третьей алкильной группы приводит к заметному снижению основности. Такая картина объясняется тем, что основность амина в воде определяется не только величиной возникающего на атоме азота отрицательного заряда, но и способностью катиона, образующегося после присоединения протона, к сольватации, и, следовательно, его стабилизацией. Чем больше атомов водорода связано с атомом азота, тем сильнее проявляется сольватация за счет возникновения межмолекулярных водородных связей и тем стабильнее становится катион. В приведенном ряду соединений основность увеличивается, но стабилизация катиона в результате гидратации в этом же направлении падает и снижает проявление основности. Подобное изменение не наблюдается, если измерения основности проводятся в растворителях, в которых водородные связи отсутствуют: основность бутиламинов в хлорбензоле возрастает в ряду: С4Н9NH2 < (С4Н9)2NH < (С4Н9)3N.
Лекция № 5
Конкурентные реакции нуклеофильного замещения и элиминирования у насыщенного атома углерода
В реакциях нуклеофильного замещения в качестве субстратов выступают спирты, тиолы, амины, галогенпроизводные, т.е. соединения, в молекулах которых содержатся sp3-гибридизированные атомы углерода, связанные ковалентной полярной связью с более электроотрицательным атомом функциональной группы. В качестве нуклеофильных частиц в этих реакциях выступают анионы и нейтральные молекулы, имеющие атом с одной или несколькими парами электронов.
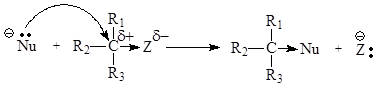
Существование большого количества потенциальных нуклеофилов (Nu) и уходящих групп (Z) позволяет широко использовать SN2 -реакции в органическом синтезе:
| Нуклеофил + | Субстрат → | Продукт + | Уходящая группа |
| I¯ + | CH3Cl ® | CH3I | + Cl¯ |
| CH3O¯ + | CH3Cl ® | CH3OCH3 | + Cl¯ |
| HS¯ + | CH3Br ® | CH3SH | + Br¯ |
| (CH3)2N- + | CH3I ® | (CH3)3N | + I¯ |
| HC≡C¯ + | CH3 –CH2 – I ® | HC≡C–CH2–CH3 | + I¯ |
| OH¯ + | CH3Br ® | CH3 – OH | + Br¯ |
| CH3–CH2–I ® | 
| + I¯ |
Реакционная способность субстрата зависит:
От полярности связи Сd+®Zd- (статический фактор). Чем выше электроотрицательность заместителя Z, тем больше полярность связи, тем больший положительный заряд возникает на атоме углерода, тем он больше предрасположен к нуклеофильной атаке.
От способности заместителя Z к отщеплению и образованию устойчивой частицы (динамический фактор). Чем длиннее связь Сd+®Zd-, тем она менее прочная и тем легче она подвергается разрыву. Длина связи Сd+®Zd- увеличивается с ростом атомного радиуса заместителя; одновременно с увеличением радиуса заместителя увеличивается также стабильность уходящей группы. Хорошо уходящими группами являются галогенид–ионы: F- < Cl- < Br- < I-. С увеличением ионных радиусов возрастает стабильность анионов, так как отрицательный заряд делокализуется в большем объеме. Плохо уходящими группами являются анионы, являющиеся сильными основаниями: Н-; ОН-; СН3О-; NH2-; СН3- и др.
Реакционная способность нуклеофильных частиц тем выше, чем легче они предоставляют пару электронов для образования связи с электронодефицитным атомом углерода. У галогенид-ионов реакционная способность в ряду F- < Cl- < Br- < I- увеличивается, что объясняется уменьшением электроотрицательности и ростом радиуса. С ростом радиуса увеличивается поляризуемость ионов, при этом пара электронов слабее удерживается ядром и легче предоставляется для образования связи с электрофильной частицей. Аналогичным образом объясняется разная реакционная способность гидроксид-ионов и гидросульфид-ионов и их аналогов: HS- > HO- ; CH3S- > CH3O-.
Нуклеофильное замещение может идти по одному из двух механизмов – SN2 или SN1. По механизму бимолекулярного нуклеофильного замещения SN2 реагируют субстраты, содержащие уходящую группу, связанную с первичным атомом углерода.
В качестве субстратов в реакциях, идущих по механизму SN2, могут выступать алкилгалогениды, спирты, тиолы, амины, содержащие первичные углеводородные радикалы.
Общая схема бимолекулярного нуклеофильного замещения SN2 :
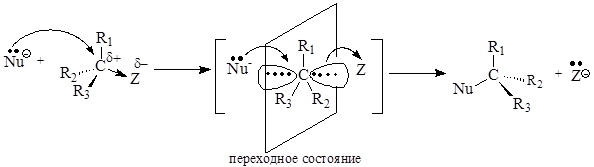
Реакции бимолекулярного нуклеофильного замещения представляют собой согласованный процесс, так как образование связи с нуклеофилом и разрыв связи с уходящей группой происходит одновременно, атакующая нуклеофильная частица постепенно вытесняет уходящую группу. При этом часть энергии, необходимая для разрыва связи С-Z, возмещается за счет энергии, выделяющейся при образовании связи С-Nu. Эти реакции являются бимолекулярным процессом, так как в образовании переходного состояния (самая медленная стадия) участвуют обе частицы и скорость реакции зависит от концентрации как нуклеофила, так и субстрата.
В переходном состоянии изменяется тетраэдрическое расположение связей атома углерода, подвергающегося нуклеофильной атаке: три заместителя R1, R2, R3 располагаются в одной плоскости, перпендикулярной плоскости рисунка, а уходящая группа и нуклеофильная частица расположены на одной линии, перпендикулярной этой плоскости. Наличие объемных заместителей R1, R2, R3, связанных с атакуемым атомом углерода, затрудняет нуклеофильную атаку, так как атаке подвергается тыльная сторона связи С-Z.
Увеличение полярности растворителя также снижает скорость реакции. Это объясняется тем, что полярный растворитель стабилизирует нуклеофильную частицу, имеющую отрицательный заряд, лучше, нежели переходное состояние, в котором отрицательный заряд распределен между нуклеофилом и уходящей группой. Это приводит к тому, что реакционная способность нуклеофила уменьшается и увеличивается разность энергий исходного вещества и переходного состояния, что ведет к увеличению энергии активации реакции.
Рассмотрим в качестве примеров реакции гидролиза галогеналканов и реакции превращения спиртов в галогеналканы.

Гидролиз галогеналканов осуществляется действием водных растворов щелочей. В качестве нуклеофильной частицы в этой реакции выступает гидроксид-ион, образующийся при диссоциации щелочи.

Однако превращение спиртов в галогенпроизводные не удается осуществить действием на спирты солями галогеноводородных кислот:

Это объясняется наличием в субстрате (спирте) плохо уходящей группы ОН-. Протекание реакции возможно при переводе плохо уходящей группы (ОН-) в хорошо уходящую группу (Н2О), что достигается действием на спирты соответствующих кислот:
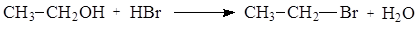
Ион водорода протонирует атом кислорода гидроксильной группы (быстрая стадия), что приводит к возникновению на атоме кислорода полного положительного заряда. Вследствие этого связь С®ОН становится более полярной, а на атоме углерода увеличивается частичный положительный заряд, что делает его более восприимчивым к нуклеофильной атаке.
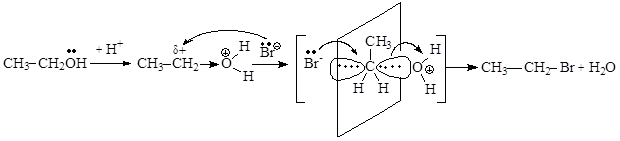
В качестве субстратов в реакциях, идущих по механизму мономолекулярного нуклеофильного замещения SN1, могут выступать алкилгалогениды, спирты, тиолы, амины, содержащие третичные углеводородные радикалы.
Общая схема мономолекулярного нуклеофильного замещения SN1:
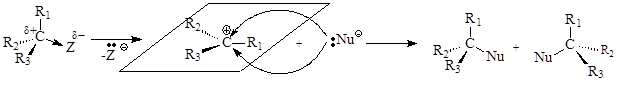
Реакции SN1 не являются согласованным процессом и состоят из двух отдельных стадий. На первой стадии происходит ионизация (медленная стадия) исходного соединения с образованием катиона и аниона. На второй стадии (быстрая стадия) образовавшийся катион реагирует с нуклеофильной частицей, давая конечный продукт. Скорость реакции в целом зависит от скорости образования катиона. Геометрия карбкатиона, отвечающая минимуму энергии, характеризуется плоским расположением связей относительно положительно заряженного sp2-гибридизированного атома углерода. Скорость взаимодействия катиона с нуклеофилом не зависит от того, с какой стороны плоскости происходит атака. В результате реакции образуется эквимолярная смесь стереоизомеров (энантиомеров). Протеканию реакций по этому механизму способствует наличие объемных заместителей у электронодефицитного атома углерода, которые затрудняют нуклеофильную атаку с тыла. Проведение реакций в полярных растворителях также способствует протеканию реакции по механизму SN1, так как под действием полярных молекул растворителя легче происходит гетеролитический разрыв связи С-Z (ионизация), а также лучше стабилизируется уходящая группа Z-. Субстраты, у которых уходящая группа связана с вторичным атомом углерода, реагируют либо по механизму SN1, либо по механизму SN2.
В качестве примера рассмотрим реакцию превращения трет-бутанола в трет-бутилбромид. Как и в случае с первичными спиртами, необходимо реакцию проводить в кислой среде для превращения плохо уходящей группы – гидроксид-иона (НО-) в хорошо уходящую группу – молекулу Н2О.
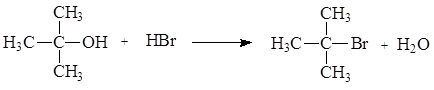
Реакция протекает в три последовательные стадии: 1) протонирование спирта – быстрая стадия; 2) гетеролитический разрыв связи С-О, приводящий к отщеплению воды и образованию карбкатиона – медленная стадия реакции, определяющая скорость реакции в целом; 3) нуклеофильная атака бромид-ионом образовавшегося на предыдущей стадии карбкатиона, в результате которой образуется конечный продукт – быстрая стадия.
Механизм реакции:
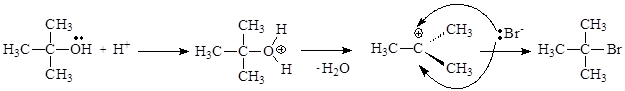
На механизм нуклеофильного замещения большое влияние оказывают заместители при электронодефицитном атоме углерода. При изучении реакций гидролиза в ряду галогеналканов
СН3Cl C6H5CH2Cl (C6H5)2CHCl (C6H5)3CCl
наблюдалось протекание реакции по SN1 механизму, начиная с бензилхлорида – C6H5CH2Cl, а ионизация трифенилхлорметана – (C6H5)3CCl - выражена настолько сильно, что растворы этого соединения в жидком SO2 обладают электропроводностью. Причиной столь сильного облегчения ионизации, и связанного с этим быстрого перехода к механизму SN1 является значительная стабилизация карбкатиона, обусловленная делокализацией положительного заряда:

При переходе от бензилхлорида к дифенилхлорметану и трифенилхлорметану эффект стабилизации карбкатиона становится все более отчетливо выраженным и атака по механизму SN1 облегчается еще сильнее, так как возможность делокализации положительного заряда возрастает. Аналогичная стабилизация наблюдается также для аллильных карбкатионов:
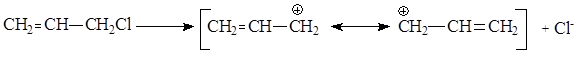
Схема реакции гидролиза бензилхлорида имеет вид:
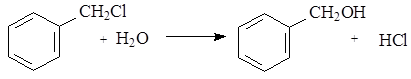
Реакция протекает в три последовательные стадии:
1) гетеролитический разрыв связи С-Сl, протекающий под действием полярных молекул растворителя – медленная стадия реакции;
2) нуклеофильная атака молекулой воды образующегося карбкатиона с образованием оксониевого катиона – быстрая стадия;
3) отщепление протона от оксониевого катиона с образованием спирта – быстрая стадия.
Механизм реакции:
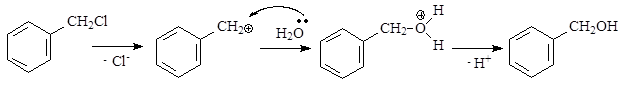
Аллилгалогениды, подобно бензилгалогенидам, обладают значительно большей реакционной способностью по сравнению с соединениями, для которых невозможна стабилизация карбкатионов за счет делокализации положительного заряда. Однако особенностью аллилгалогенидов и бензилгалогенидов является то, что группа СН2 в этих соединениях значительно легче атакуется также по механизму SN2, чем группа СН2 в хлорэтане (замещение идет » в 100 раз быстрее). Такой эффект объясняется тем, что sp2-гибридизированный атом углерода в переходном SN2 состоянии использует свою негибридную р-орбиталь для взаимодействия не только с атакующей нуклеофильной частицей и отщепляющейся уходящей группой, но также с p-орбиталью двойной связи, что приводит к стабилизации переходного состояния.
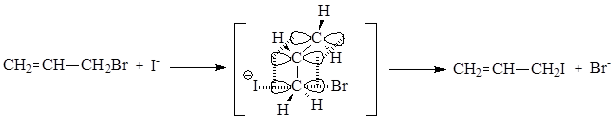
Рассмотрев оба механизма нуклеофильного замещения, можно сделать некоторые обобщения о влиянии различных факторов на механизм реакции:
Реакция протекает по SN1 механизму в случае тех соединений, которые способны образовывать достаточно стабильные катионы. Триалкилметильные и мезомерно стабилизированные катионы обеспечивают преимущественное протекание реакции мономолекулярного замещения. Увеличение числа алкильных заместителей облегчает мономолекулярное и затрудняет бимолекулярное замещение.
Растворители с высокой диэлектрической проницаемостью (Н2О, НСООН, водноорганические смеси) стабилизируют ионы и поэтому способствуют мономолекулярному замещению, растворители с низкой диэлектрической проницаемостью (диметилформамид), не вызывающие стабилизации ионов, способствуют бимолекулярному замещению.
Дата добавления: 2015-08-08; просмотров: 6162;