Особенности ферментативного катализа. 2 страница
ваться на точном знании механизма действия лекарственных средств на
биосинтез ферментов, на активность уже синтезированных ферментов или
на регуляцию их активности в организме. Иногда для лечения некоторых
болезней используют избирательно действующие ингибиторы. Так, инги-
битор ряда протеиназ (трипсина, химотрипсина и калликреина) трасилол
широко применяется для лечения острого панкреатита – болезни, при ко-
торой уровень трипсина и химотрипсина в крови резко возрастает. Знание
избирательного ингибиторного действия некоторых природных и синте-
тических соединений (так называемых антиметаболитов) на ферменты
может служить методологической основой для разработки эффективных
методов синтеза химиотерапевтических препаратов. Этот путь открывает
широкие возможности для направленного воздействия на синтез ферментов
в организме и регуляции интенсивности метаболизма при патологии.
Типы ингибирования. Различают обратимое и необратимое ингибиро-
вание. Если ингибитор вызывает стойкие изменения пространственной
третичной структуры молекулы фермента или модификацию функциональ-
ных групп фермента, то такой тип ингибирования называется необрати-
мым. Чаще, однако, имеет место обратимое ингибирование, под-
дающееся количественному изучению на основе уравнения Михаэлиса-
Ментен. Обратимое ингибирование в свою очередь разделяют на кон-
курентное и неконкурентное в зависимости от того, удается или не удается
преодолеть торможение ферментативной реакции путем увеличения кон-
центрации субстрата.
Конкурентное ингибирование.
Неконкурентное ингибирование вызывается веществами, не
имеющими структурного сходства с субстратами и часто связывающимися
не с активным центром, а в другом месте молекулы фермента. Степень
торможения во многих случаях определяется продолжительностью дейст-
вия ингибитора на фермент. При данном типе ингибирования благодаря
образованию стабильной ковалентной связи фермент часто подвергается
полной инактивации, и тогда торможение становится необратимым. При-
мером необратимого ингибирования является действие йодацетата, ДФФ,
а также диэтил-n-нитрофенилфосфата и солей синильной кислоты. Это
действие заключается в связывании и выключении функциональных групп
или ионов металлов и молекуле фермента.
Следует указать, что неконкурентное ингибирование также может быть
обратимым и необратимым, поскольку отсутствует конкуренция между
субстратом и ингибитором за активный центр. Примеры необратимого
ингибирования приведены ранее. При обратимом неконкурентном
ингибировании субстрат S и ингибитор I связываются с разными
центрами, поэтому появляется возможность образования как комплекса EI,
так и тройного комплекса EIS; последний может распадаться с осво-
бождением продукта, но с меньшей скоростью, чем комплекс ES.
Этот тип неконкурентного ингибирования чаще всего наблюдается
у ферментов, катализирующих превращения более одного субстрата, когда
связывание ингибитора не блокирует связывание субстрата с активным
центром. Ингибитор при этом соединяется как со свободным ферментом,
так и с ES-комплексом.
Известно, кроме того, так называемое бесконкурентное ингиби-
рование, когда ингибитор связывается с ферментом также в некатали-
тическом центре, однако не со свободным ферментом, а только с ES-комп-
лексом в виде тройного комплекса.
Для выяснения вопроса о типе ингибирования пользуются уравнениями
Михаэлиса-Ментен, Лайнуивера-Бэрка или другими, например уравне-
нием Эди-Хофсти:
v = –Km(v/[S]) + Vmax
и соответствующими графиками в прямолинейных координатах.
При конкурентном типе ингибирования ингибитор увеличивает значение
Кm, не оказывая влияния на максимальную скорость Vmax (рис. 4.21). Это
означает, что при достаточно высокой концентрации субстрата [S] ин-
гибитор вытесняется молекулами субстрата из комплекса EI. При некон-
курентном ингибировании (рис. 4.22) ингибитор снижает величину макси-
мальной скорости. Если при этом величина Кm не уменьшается, то говорят
о полностью неконкурентном ингибировании. Подобный тип ингибиро-
вания имеет место при образовании неактивных, труднодиссоциирующих
комплексов EI и(или) EIS. Часто, однако, наблюдается смешанный тип
ингибирования, иногда называемый частично неконкурентным, или обра-
тимым неконкурентным ингибированием (см. ранее), при котором сни-
жение Vmax сочетается с одновременным увеличением значений Кm. Это
означает, что комплекс EI сохраняет частичную активность, т.е. способ-
ность к образованию промежуточного тройного комплекса EIS, в котором
субстрат подвергается
13.
Конкурентное ингибирование может быть вызвано веществами,
имеющими структуру, похожую на структуру субстрата, но несколько
отличающуюся от структуры истинного субстрата. Такое ингибирование
основано на связывании ингибитора с субстратсвязывающим (активным)
центром. Классическим примером подобного типа ингибирования является
торможение сукцинатдегидрогеназы (СДГ) малоновой кислотой. Этот фер-
мент катализирует окисление путем дегидрирования янтарной кислоты
(сукцината) в фумаровую:
Если в среду добавить малонат (ингибитор), то в результате структур-
ного сходства его с истинным субстратом сукцинатом (наличие двух таких
же ионизированных карбоксильных групп) он будет взаимодействовать
с активным центром с образованием фермент-ингибиторного комплекса,
однако при этом полностью исключается перенос атома водорода от
малоната. Структуры субстрата (сукцинат) и ингибитора (малонат) все же
несколько различаются. Поэтому они конкурируют за связывание с актив-
ным центром, и степень торможения будет определяться соотношением
концентраций малоната и сукцината, а не абсолютной концентрацией
ингибитора. Таким образом, ингибитор может обратимо связываться
с ферментом, образуя фермент-ингибиторный комплекс. Этот тип ингиби-
рования иногда называют ингибированием по типу метаболического анта-
гонизма .
В общей форме реакция взаимодействия ингибитора с ферментом может
быть представлена следующим уравнением:
Образовавшийся комплекс, называемый фермент-ингибиторным комп-
лексом ЕI, в отличие от фермент-субстратного комплекса ES не распадается
с образованием продуктов реакции. Константу диссоциации комплекса EI,
или ингибиторную константу Кi, можно, следуя теории Михаэлиса–Мен-
тен, определить как отношение констант обратной и прямой реакций:
т.е. ингибиторная константа прямо пропорциональна произведению кон-
центрации фермента и ингибитора и обратно пропорциональна концент-
рации комплекса EI.
Метод конкурентного торможения нашел широкое применение в ме-
дицинской практике. Известно, например, что для лечения некоторых
инфекционных заболеваний, вызываемых бактериями, применяют сульфа-
ниламидные препараты. Оказалось, что эти препараты имеют структурное
сходство с парааминобензойной кислотой, которую бактериальная клетка
использует для синтеза фолиевой кислоты, являющейся составной частью
ферментов бактерий. Благодаря этому структурному сходству сульфани-
ламид блокирует действие фермента путем вытеснения парааминобензой-
ной кислоты из комплекса с ферментом, синтезирующим фолиевую кисло-
ту, что ведет к торможению роста бактерий.
Некоторые аналоги витамина В6 и фолиевой кислоты, в частности
дезоксипиридоксин и аминоптерин (см. главу 7), действуют как конкурент-
ные, так называемые коферментные, ингибиторы (или антивитамины),
тормозящие многие интенсивно протекающие при патологии биологи-
ческие процессы в организме. Применение подобных аналогов в меди-
цинской практике (в частности, в дерматологии и онкологии) основано на
конкурентном вытеснении коферментов из субстратсвязывающих центров
ключевых ферментов обмена.
14.неконкурентное
Неконкурентное ингибирование вызывается веществами, не
имеющими структурного сходства с субстратами и часто связывающимися
не с активным центром, а в другом месте молекулы фермента. Степень
торможения во многих случаях определяется продолжительностью дейст-
вия ингибитора на фермент. При данном типе ингибирования благодаря
образованию стабильной ковалентной связи фермент часто подвергается
полной инактивации, и тогда торможение становится необратимым. При-
мером необратимого ингибирования является действие йодацетата, ДФФ,
а также диэтил-n-нитрофенилфосфата и солей синильной кислоты. Это
действие заключается в связывании и выключении функциональных групп
или ионов металлов и молекуле фермента.
Следует указать, что неконкурентное ингибирование также может быть
обратимым и необратимым, поскольку отсутствует конкуренция между
субстратом и ингибитором за активный центр. Примеры необратимого
ингибирования приведены ранее. При обратимом неконкурентном
ингибировании субстрат S и ингибитор I связываются с разными
центрами, поэтому появляется возможность образования как комплекса EI,
так и тройного комплекса EIS; последний может распадаться с осво-
бождением продукта, но с меньшей скоростью, чем комплекс ES.
Е + S àESàE+P
+ +
I I
ЕI+ SàESI
Этот тип неконкурентного ингибирования чаще всего наблюдается
у ферментов, катализирующих превращения более одного субстрата, когда
связывание ингибитора не блокирует связывание субстрата с активным
центром. Ингибитор при этом соединяется как со свободным ферментом, так и с ES комплексом.
15.Ингибиторы-лекарства,
Лекарственные препараты, применяемые с целью подавления активности ферментов, называются ингибиторами ферментов.
Классификация
1. Ингибиторы протеиназ: контрикал.
2. Ингибиторы фибринолиза: кислота амино-капроновая.
3. Антихолинэстеразные средства: прозерин, физостигмина салицилат, галантамина гидробромид и др.
4. Ингибиторы МАО: ниаламид.
5. Ингибиторы карбоангидразы: диакарб.
6. Ингибиторы ксантиноксидазы: аллопуринол.
7. Ингибиторы ацетальдегидрогеназы: циамид, тетурам (дисульфирам) и др.
Контрикал — антиферментный препарат, ингибирующий активность трипсина, калликреина, плазмина.
Фармакокинетика: при внутривенном введении действие развивается через 10—15 мин.
Показания к применению: острый панкреатит, панкреанекроз в сочетании с гепарином в острый период инфаркта миокарда.
Противопоказания: с осторожностью у лиц, склонных к аллергическим реакциям.
Побочные эффекты: аллергические реакции.
16. Под коферментом часто подразумевают дополнительную группу,
легко отделяемую от апофермента при диссоциации. Предполагают, что
простетическая группа может быть связана с белком ковалентными и неко-
валентными связями. Так, в молекуле ацетилкоэнзим-А-карбоксилазы ко-
фактор биотин ковалентно связан с апоферментом посредством амидной
связи (см. главу 7). С другой стороны, химические связи между кофакто-
рами и пептидными цепями могут быть относительно слабыми (например,
водородные связи, электростатические взаимодействия и др.). В таких
случаях при выделении ферментов наблюдается полная диссоциация обеих
частей, и изолированый белковый компонент оказывается лишенным фер-
ментативной активности, пока не будет добавлен извне недостающий
кофактор. Именно к подобным изолированным низкомолекулярным орга-
ническим веществам применим термин «кофермент», типичными предста-
вителями которых являются витамины В1, В2, В6, РР, содержащие кофер-
менты. Известно также, что и простетические группы, и коферменты
активно включаются в химические реакции, выполняя функции промежу-
тоных переносчиков электронов, атомов водорода или различных функцио-
нальных групп (например, аминных, ацетильных, карбоксильных). В подоб-
ных случаях кофермент рассматривают в качестве второго субстрата, или
косубстрата.
Роль кофермента (Ко) в качестве переносчика, например, атомов водо-
рода может быть представлена в виде схемы, где SH – субстрат, КоЕ – хо-
лофермент, А – акцептор протона:
SHКoЕ AH
S КоЕН A
Субстрат подвергается окислению, отдавая электроны и протоны,
а КоЕ – восстановлению, принимая электроны и протоны. В следующей
полуреакции восстановленный КоЕН может отдавать электроны и протоны
на какой-либо другой промежуточный переносчик электронов и протонов
или на конечный акцептор (см. главу 9).
Коэнзим, кофактор, простетическая группа – двусмысленный биохими-
ческий жаргон. До сих пор продолжается терминологический спор, по-
скольку часто определения «коэнзим», «кофактор» и «простетическая груп-
па» рассматриваются через призму их роли в реакциях энзиматического
(ферментативного) катализа. Следует, однако, считаться с тем неоспо-
римым фактом, что во многих случаях небелковые органические молекулы,
как и ионы металлов, абсолютно необходимы белковому компоненту при
выполнении определенной биологической функции, не имеющей отношения
к биокатализу. Несомненно, имеют значение также тип и характер связи
небелкового компонента с молекулой белка. Поэтому очевидно, что ко-
фактором может служить любой фактор, абсолютно необходимый для
выполнения белком его каталитической или любой другой биологической
роли. С другой стороны, коферментом может быть любой небелковый
фактор, который непосредственно вовлечен в реакцию энзиматического
катализа. Кофактор, который непосредственно не участвует в акте ката-
лиза, не является коэнзимом. В то же время простетическую группу
(ковалентно связанный небелковый компонент, необходимый для опреде-
ленной функции) можно назвать коферментом, если она непосредственно
участвует в энзиматической реакции. Простетическая группа, которая не
вовлечена в акт катализа, но функционально является существенным как
для фермента, так и для некаталитического белка, может быть названа
кофактором. И наконец, кофактор и кофермент, непрочно связанные (или
слабо связанные) с ферментом или белком, тем не менее не классифи-
цируются в качестве простетических групп.
Многие двухвалентные металлы (Mg2+, Мn2+, Са2+), как будет пока-
зано далее, также выполняют роль кофакторов, хотя они не относятся ни
к коферментам, ни к простетическим группам. Известны примеры, когда
ионы металлов прочно связаны с белковой молекулой, выполняя функции
простетической группы. В частности, очищенный фермент, катализирую-
щий окисление аскорбиновой кислоты (витамин С) в дезоксиаскорбиновую
кислоту, содержит 8 атомов меди на одну молекулу; все они настолько
прочно связаны с белковой молекулой, что даже не обмениваются с ионо-
обменными смолами и не отделяются методом диализа. Более того,
с помощью метода электронного парамагнитного резонанса показано
участие ионов меди в промежуточном переносе электронов. Интересно
отметить, что свободные ионы меди также наделены каталитической
активностью при окислении аскорбиновой кислоты, однако эта активность
повышается во многие тысячи раз, если ионы меди соединяются с апофер-
ментом в единый комплекс – холофермент.
Данные о важнейших коферментах и простетических группах ферментов,
включая их наименования и структуру, химическую природу витамина,
входящего в их состав, и характер выполняемой биохимической функции
в метаболизме, детально рассмотрены в главах 7 и 9–13.
Получены доказательства кофакторной функции в ферментативных
реакциях и ряда других биологически активных соединений, не относящихся
к витаминам: HS-глутатиона, АТФ, липоевой кислоты, производных ну-
клеозидов (уридинфосфат, цитидинфосфат, фосфоаденозинфосфосульфат),
порфиринсодержащих веществ и др. Сюда же могут быть отнесены тРНК,
которые в составе ферментов аминоацил-тРНК-синтетаз принимают ак-
тивное участие в транспорте аминокислот в рибосоме, где осуществляется
синтез белка (см. главу 14).
Следует отметить одну отличительную особенность двухкомпонентных
ферментов: ни кофактор отдельно (включая большинство коферментов), ни
сам по себе апофермент каталитической активностью не наделены, и только
их объединение в одно целое, протекающее не хаотично, а в соответствии
с программой их структурной организации, обеспечивает быстрое про-
текание химической реакции.
17.Коферменты:
По химической природе: витаминные, витаминоподобные, невитаминные.
По механизму действия: переносчики атомов водорода,электронов,протонов.
переносчики отдельных химических групп.
18.Над и НАДФ
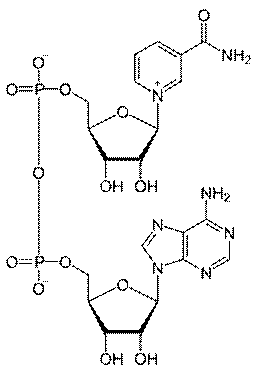
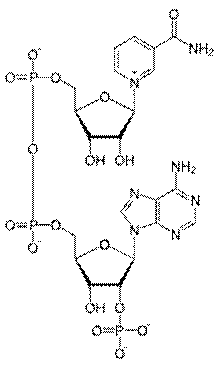
Производные PP витамина. Биохимическая функция: дыхание,перенос водорода.
Витамин РР входит в состав НАД или НАДФ,
являющихся коферментами большого числа обратимо действующих в
окислительно-восстановительных реакциях дегидрогеназ (формулы ко-
ферментов приведены в главе 9).
Показано, что ряд дегидрогеназ использует только НАД и НАДФ
(соответственно малатдегидрогеназа и глюкозо-6-фосфатдегидрогеназа),
другие могут катализировать окислительно-восстановительные реакции
в присутствии любого из них (например, глутаматдегидрогеназа; см. главу
12). В процессе биологического окисления НАД и НАДФ выполняют роль
промежуточных переносчиков электронов и протонов между окисляемым
субстратом и флавиновыми ферментами (молекулярные механизмы учас-
тия пиридиновых нуклеотидов в этом процессе подробно рассматриваются
19.ФАД и ФМН


Производные витамина B2.Биологическая функция дыхание, перенос водорода.
Рибофлавин входит в состав флавиновых кофер-
ментов, в частности ФМН и ФАД *, являющихся в свою очередь просте-
тическими группами ферментов ряда других сложных белков – флаво-
протеинов. Некоторые флавопротеины в дополнение к ФМН или ФАД
содержат еще прочно связанные неорганические ионы, в частности железо
или молибден, наделенные способностью катализировать транспорт элек-
тронов. Различают 2 типа химических реакций, катализируемых этими
ферментами. К первому относятся реакции, в которых фермент осуществ-
ляет прямое окисление с участием кислорода, т.е. дегидрирование (от-
щепление электронов и протонов) исходного субстрата или промежуточ-
ного метаболита. К ферментам этой группы относятся оксидазы L- и
D-аминокислот, глициноксидаза, альдегидоксидаза, ксантиноксидаза и др.
Вторая группа реакций, катализируемых флавопротеинами, характеризует-
ся переносом электронов и протонов не от исходного субстрата, а от
восстановленных пиридиновых коферментов. Ферменты этой группы иг-
рают главную роль в биологическом окислении. В каталитическом цикле
изоаллоксазиновый остаток ФАД или ФМН подвергается обратимому
восстановлению с присоединением электронов и атомов водорода к N1
и N10. ФМН и ФАД прочно связываются с белковым компонентом, иногда
даже ковалентно, как, например, в молекуле сукцинатдегидрогеназы.
ФМН синтезируется в организме животных из свободного рибофлавина
и АТФ при участии специфического фермента рибофлавинкиназы:
Образование ФАД в тканях также протекает при участии специфического
АТФ-зависимого фермента ФМН-аденилилтрансферазы. Исходным ве-
ществом для синтеза является ФМН:
20.КоА
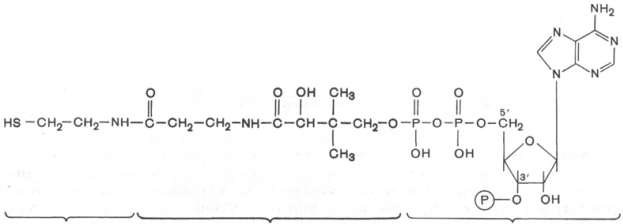
Витамин В3,транспорт ацильных групп.
Пантотеновая кислота входит в состав кофер-
мента А, или коэнзима А (КоА). Название «коэнзим А» (кофермент
ацилирования) связано с тем, что это соединение участвует в фермен-
тативных реакциях, катализирующих как активирование, так и перенос
ацетильного радикала СН3СО; позже оказалось, что КоА активирует
и переносит также другие кислотные остатки (ацилы). В результате обра-
зования ацил-КоА происходит активация карбоновой кислоты, которая
поднимается на более высокий энергетический уровень, создающий вы-
годные термодинамические предпосылки для ее использования в реакциях,
протекающих с потреблением энергии.
Строение КоА расшифровал Ф. Линен. В основе структуры лежит
остаток 3'-фосфоаденозин-5'-дифосфата (отличается от АТФ наличием у
3'-гидроксила фосфатной группы), соединенный с остатком пантотеновой
кислоты, карбонильная группа которой в свою очередь связана с остатком
β-меркаптоэтиламина (тиоэтиламина).
Реакционноспособным участком молекулы КоА в биохимических реак-
циях является SH-группа, поэтому принято сокращенное обозначение КоА
в виде SH-KoA. О важнейшем значении КоА в обмене веществ (как будет
показано далее – см. главы 9–11) свидетельствуют обязательное непосред-
ственное участие его в основных биохимических процессах, окисление
и биосинтез высших жирных кислот, окислительное декарбоксилирование
α-кетокислот (пируват, α-кетоглутарат), биосинтез нейтральных жиров,
фосфолипидов, стероидных гормонов, гема гемоглобина, ацетилхолина,
гиппуровой кислоты и др.
21.Тиаминпирофосфат
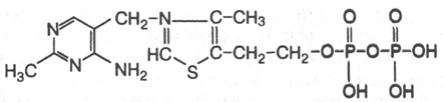
В1, декарбоксилирование а-кетокислот,перенос активного альдегида (транскетолаза).
Экспериментально доказано, что витамин B1 в
форме ТПФ является составной часть минимум 5 ферментов, участвующих
в промежуточном обмене веществ. ТПФ входит в состав двух сложных
ферментных систем – пируват- и α-кетоглутаратдегидрогеназных
комплексов, катализирующих окислительное декарбоксилирование
пировиноградной и α-кетоглутаровой кислот. В составе транскетолазы
ТПФ участвует в переносе гликоальдегидного радикала от кетосахаров на
альдосахара (см. главу 10). ТПФ является коферментом пируватдекар-
боксилазы клеток дрожжей (при алкогольной ферментации) и дегидро-
геназы γ-оксикетоглутаровой кислоты.
Приведенными примерами, вероятнее всего, не ограничиваются биоло-
гические функции тиамина. В частности, ТПФ участвует в окислительном
декарбоксилировании глиоксиловой кислоты и α-кетокислот, образующих-
ся при распаде аминокислот с разветвленной боковой цепью; в растениях
ТПФ является эссенциальным кофактором при синтезе валина и лейцина
в составе фермента ацетолактатсинтетазы.
22.Пиродаксальфосфат.
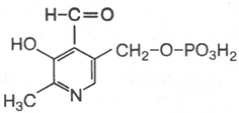
В6,обмена аминокислот,перенос аминогрупп.
Оказалось, что, хотя все три производных 3-окси-
пиридина наделены витаминными свойствами, коферментные функции
выполняют только фосфорилированные производные пиридоксаля и пи-
ридоксамина.
Фосфорилирование пиридоксаля и пиридоксамина является фермен-
тативной реакцией, протекающей при участии специфических киназ. Синтез
пиридоксальфосфата, например, катализирует пиридоксалькиназа, которая
наиболее активна в ткани мозга. Эту реакцию можно представить сле-
дующим уравнением:
Пиридоксаль + АТФ –> Пиридоксальфосфат + АДФ.
Доказано, что в животных тканях происходят взаимопревращения
пиридоксальфосфата и пиридоксаминфосфата, в частности в реакциях
трансаминирования и декарбоксилирования аминокислот (см. главу 12).
Следует отметить, что в выяснение биологической роли витамина В6
и пиридоксальфосфата в азотистом обмене существенный вклад внесли
А.Е. Браунштейн, С.Р. Мардашев, Э. Снелл, Д. Мецлер, А. Майстер и др.
Известно более 20 пиридоксалевых ферментов, катализирующих ключевые
реакции азотистого метаболизма во всех живых организмах. Так доказано,
что пиридоксальфосфат является простетической группой аминотранс-
фераз, катализирующих обратимый перенос аминогруппы (NH2-группы)
от аминокислот на α-кетокислоту, и декарбоксилаз аминокислот, осу-
ществляющих необратимое отщепление СО2 от карбоксильной группы
аминокислот с образованием биогенных аминов. Установлена кофер-
ментная роль пиридоксальфосфата в ферментативных реакциях неокисли-
тельного дезаминирования серина и треонина, окисления триптофана,
кинуренина, превращения серосодержащих аминокислот, взаимопревраще-
ния серина и глицина (см. главу 12), а также в синтезе δ-аминолевулиновой
кислоты, являющейся предшественником молекулы гема гемоглобина, и др.
Пиридоксин относится к витаминам, коферментная роль которых изучена
наиболее подробно. В последние годы число вновь открытых пиридокса-
левых ферментов быстро увеличивалось. Так, для действия гликогенфос-
форилазы существенной оказалась фосфорильная, а не альдегидная группа
пиридоксальфосфата. Вследствие широкого участия пиридоксальфосфата
в процессах обмена при недостаточности витамина В6 отмечаются разно-
образные нарушения метаболизма аминокислот.
23.Тетрагидрофолиевая кислота.
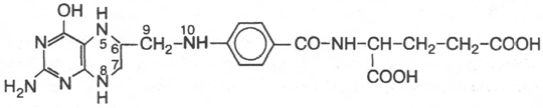
Фолиевая кислота,Транспорт одноуглеродных групп.
Коферментные функции фолиевой кислоты связаны
не со свободной формой витамина, а с восстановленным его птеридиновым
производным. Восстановление сводится к разрыву двух двойных связей
и присоединению четырех водородных атомов в положениях 5, 6, 7 и 8
с образованием тетрагидрофолиевой кислоты (ТГФК). Оно протекает
в 2 стадии в животных тканях при участии специфических ферментов,
содержащих восстановленный НАДФ. Сначала при действии фолатредук-
тазы образуется дигидрофолиевая кислота (ДГФК), которая при участии
второго фермента – дигидрофолатредуктазы – восстанавливается в ТГФК:
ФК + НАДФН + Н+ <=> ДГФК + НАДФ+;
ДГФК + НАДФН + Н+ <=> ТГФК + НАДФ+
Доказано, что коферментные функции ТГФК непосредственно связаны
с переносом одноуглеродных групп, первичными источниками которых
в организме являются β-углеродный атом серина, α-углеродный атом
Фолиевая (птероилглутаминовая) кислота
5,6,7,8-Тетрагидрофолиевая кислота (ТГФК)
глицина, углерод метальных групп метионина, холина, 2-й углеродный
атом индольного кольца триптофана, 2-й углеродный атом имидазольного
кольца гистидина, а также формальдегид, муравьиная кислота и метанол.
К настоящему времени открыто шесть одноуглеродных групп, включа-
ющихся в разнообразные биохимические превращения в составе ТГФК:
формильная (—СНО), метильная (—СН3), метиленовая (—СН2—),
метенильная (—СН=), оксиметильная (—СН2ОН) и формими-
новая (—CH=NH). Выяснено, что присоединение этих фрагментов к
ТГФК является ферментативной реакцией ковалентного связывания их
с 5-м или 10-м атомом азота (или с обоими атомами вместе). В качестве
примера приводим отдельные функциональные группы в активных участках
ТГФК:
Имеются данные, что производные ТГФК участвуют в переносе одно-
углеродных фрагментов при биосинтезе метионина и тимина (перенос
метильной группы), серина (перенос оксиметильной группы), образовании
пуриновых нуклеотидов (перенос формильной группы) и т.д. (см. главы 12
и 13). Перечисленные вещества играют исключительно важную, ключевую,
Дата добавления: 2015-04-05; просмотров: 2201;