Патогенез диабетического кетоацидоза
Ведущие патогенетические звенья диабетического кетоацидоза, учитывающие нарушения всех видов обмена, представлены на интегральной схеме (рис. 12-28).
Среди важнейших метаболических нарушений при СД выделяются следующие:
1. Нарушения углеводного обменапри отсутствии инсулина (или его действия) характеризуются гипергликемией, глюкозурией и гиперлактатацидемией.
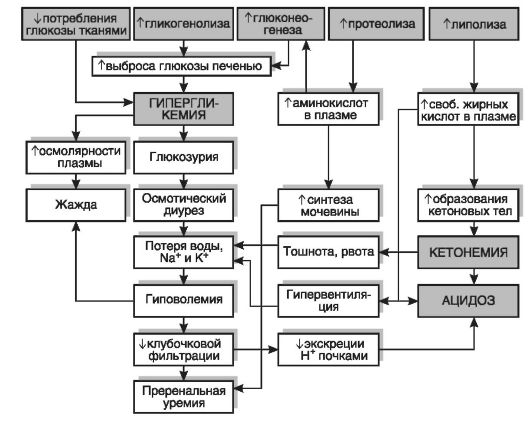 Рис. 12-28.Патогенез диабетического кетоацидоза. Верхний ряд показывает основные метаболические эффекты при недостатке инсулина, которые вызывают все последующие изменения
Рис. 12-28.Патогенез диабетического кетоацидоза. Верхний ряд показывает основные метаболические эффекты при недостатке инсулина, которые вызывают все последующие изменения
Гипергликемияобусловлена:
• нарушением поступления глюкозы из крови внутрь клеток;
• компенсаторным ускорением гликогенолиза;
• активацией глюконеогенеза вследствие снятия репрессивного действия инсулина на синтез ключевых ферментов этого метаболического пути;
• усилением секреции глюкокортикоидов, являющихся стимуляторами синтеза ключевых ферментов глюконеогенеза в печени и почках.
Глюкозурияи, как следствие, полиурияразвиваются при достижении концентрации глюкозы в крови 9,9 ммоль/л, когда преодолевается почечный порог, и глюкоза появляется в моче.
Гиперлактатацидемияразвивается вследствие торможения катаболизма лактата в ЦТК и нарушения ресинтеза пирувата из лактата (см. рис. 12-17).
2. Нарушения липидного обменапри отсутствии инсулина характеризуются усилением липолиза и снижением липогенеза. Дефицит инсулина при сахарном диабете приводит к тому, что:
а) контринсулярные гормоны (глюкагон, адреналин, глюкокортикоиды и др.) стимулируют мобилизацию липидов из жировых депо и доставку жирных кислот к органам, что является адаптивным механизмом, поставляющим альтернативный субстрат окисления в условиях снижения утилизации глюкозы клетками;
б) снижается активность ЛП-липазы адипоцитов, поэтому свободные жирные кислоты не поступают в жировую ткань;
в) начинает преобладать эффект глюкагона, стимулирующий кетогенез в печени и гормончувствительную ТАГ-липазу в адипоцитах;
г) в норме кетоновые тела стимулируют выход инсулина из поджелудочной железы, что угнетает липолиз и таким образом ограничивает доставку липидов в печень и соответственно кетогенез (при сахарном диабете этот регуляторный механизм нарушен: идет усиленная продукция кетоновых тел печенью благодаря интенсивному β-окислению жирных кислот);
д) при сахарном диабете в избыточном количестве образуется продукт β-окисления жирных кислот - ацетил-КоА, однако способность цикла Кребса утилизировать данный продукт существенно снижена. В результате этого избыток ацетил-КоА становится источником образования больших количеств кетоновых тел: β-оксимасляной, ацетоуксусной кислот и ацетона, что и приводит к кетонемии.Кетоновые тела начинают выделяться с мочой в виде натриевых солей (кетонурия),а ацетон - также и в составе выдыхаемого воздуха.
3. Нарушения белкового обменапри отсутствии инсулина характеризуются преобладанием процессов катаболизма вследствие активации глюконеогенеза из глюкогенных аминокислот и снижения проницаемости клеточных мембран для аминокислот, что приводит к недостатку в тканях свободных аминокислот и нарушению процесса синтеза белка. Стимулируется синтез мочевины, что характеризуется гиперазотемиейи приводит к отрицательному азотистому балансу.
4. Нарушения кислотно-щелочного баланса организмаразвиваются в связи с накоплением кислых продуктов метаболизма в результате переключения аэробных путей утилизации глюкозы на анаэробный гликолиз с повышенной продукцией лактата и усиления ке-
тогенеза. По мере истощения емкости буферных систем организма формируется декомпенсированный метаболический ацидоз.
5. Водно-солевой обмен и дегидратация клеток.Нарушения водного обмена при СД проявляются полиурией(суточный диурез достигает 4-10 л) и полидипсией(до 9 л воды в сутки), обусловленной гипогидратацией организма и гиперосмией крови в связи с гипергликемией, гиперазотемией, кетонемией, гиперлактатацидемией, повышением содержания отдельных ионов. Именно эти явления дали основания для первоначального названия заболевания: diabetes mellitus (лат.) - сахарное мочеизнурение.Повышение осмотического давления крови сопровождается дегидратацией клеток, особенно чувствительны к этому клетки нервной ткани.
Необходимо особо отметить, что сочетание ацидоза и явлений дегидратации в эритроцитахприводит к снижению в этих клетках концентрации 2,3-дифосфоглицериновой кислоты- аллостерического модулятора функций гемоглобина. В этих условиях сродство гемоглобина к кислороду возрастает, но его способность отдавать кислород тканям уменьшается, вследствие чего гипоксия, вызванная глубокими метаболическими нарушениями при СД, усугубляется.
В результате перечисленных выше нарушений обмена веществ при СД могут развиваться серьезные осложнения, к которым относят комы и диабетические микро- и макроангиопатии (нефропатия, слепота, синдром «диабетической стопы»), вторичные инфекции из-за иммунодефицита.
Диабетическая кома.Критическая дегидратация тканей организма с поражением функций головного мозга ведет к развитию диабетической (гипергликемической) комы.Кома развивается при достижении концентрации глюкозы в крови от 19,4 до 33,3 ммоль/л и более. В этих условиях вследствие кетоацидоза ионы калия выходят во внеклеточное пространство (гиперкалиемия), что лежит в основе нарушения сократительной функции миокарда, а также дыхательной мускулатуры. Диабетическая кома может привести к летальному исходу, если больному не будет своевременно проведена специфическая противокоматозная терапия.
Различают следующие виды диабетической комы:
1. Гипергликемическая кетоацидотическая кома.Развивается чаще всего у больных СД 1 типа вследствие гипергликемии, гиперкетонемии и метаболического ацидоза. Глюкоза и кетоновые тела выводятся с мочой (глюкозурия и кетонурия), что способ-
ствует увеличению осмотического давления в первичной моче, потере ионов Na и сопровождается полиурией. При этом возникает обезвоживание, которое ведет к недостаточности периферического кровообращения и гипоксии тканей. Ацидоз вызывает дыхание Куссмауля, при котором теряется СО2 и как следствие усугубляются нарушения водно-электролитного баланса, кислотно-щелочного равновесия, возникает резкое нарушение метаболизма и функций клеток ЦНС, что приводит к расстройству высшей нервной деятельности. К клиническим проявлениям комы относятся: слабость, головная боль, адинамия, диспепсические расстройства (в 30-50% случаев - «абдоминальный синдром» - клиника «острого живота»), дыхание Куссмауля с запахом ацетона в выдыхаемом воздухе, снижение кровяного давления и частый слабый пульс, нарастающая сухость кожи и слизистых оболочек. Затем наступает полная потеря сознания, расслабление мышц, зрачки сужаются, отмечаются характерные признаки энцефалопатии. Содержание глюкозы в крови превышает 22 ммоль/л, кетоновых тел - 17 ммоль/л, повышено содержание остаточного азота, мочевины, холестерина, жирных кислот, уровень натрия чаще нормальный, реже - снижен, уровень калия чаще нормальный, у больных с почечной недостаточностью может быть повышен.
2. Гипергликемическая гиперосмолярная кома.Встречается реже, чем кетоацидотическая, и развивается у больных СД 2 типа старше 50 лет при дополнительном воздействии обезвоживающих факторов (рвота, понос, ограничение приема жидкости, ожоги, кровопотеря, полиурия, прием диуретиков). Основными звеньями патогенеза этого вида комы являются дегидратация организма и развитие гиперосмолярности плазмы, уровень гликемии может достигать 55 ммоль/л. У больных нет выраженной гиперкетонемии и кетонурии, отсутствует запах ацетона изо рта и, если не обратиться к врачу, нарастает уровень глюкозы в крови до крайне высокой степени, что способствует усилению диуреза (глюкозурический осмотический диурез). Возникающее обезвоживание приводит к гиповолемии, стимуляции секреции альдостерона и задержке ионов Na и Cl. Показатель осмолярности плазмы повышается в 1,5-2 раза (в норме около 300 мосмоль/л, при коме достигает 500 мосмоль/л), что приводит к резко выраженной внутриклеточной дегидратации, нарушению водного и электролитного равновесия в клетках мозга, гипоксии ЦНС с выраженной неврологической симптоматикой и потере сознания.
3. Гипергликемическая кома с лактат-ацидозом (лактацидотическая).Это относительно редкое, но опасное осложнение СД. В механизме ее развития важную роль играют следующие факторы:
а) снижение активности ферментативного пируватдегидрогеназного комплекса (выявляется при дефиците инсулина), превращающего пируват в ацетил-КоА. Пируват в обратимой реакции, катализируемой лактатдегдрогеназой, превращается в молочную кислоту (см. рис. 12-17);
б) применение лекарственных препаратов, стимулирующих анаэробный гликолиз и тем самым повышающих содержание лактата и пирувата в организме (например, бигуаниды, повышающие утилизацию глюкозы за счет ее анаэробного распада). При поражении печени или почек может иметь место кумуляция этих препаратов в организме, в результате чего развиваются лактоацидоз и кома;
в) гипоксическое состояние (при котором, как правило, стимулируется гликолиз), вызванное физическим переутомлением, сердечной или дыхательной недостаточностью.
Как следствие в крови накапливается молочная кислота (содержание лактата в плазме превышает 5 ммоль/л), что сопровождается развитием коллапса, нарушением сердечной деятельности и функций дыхательного центра (возникает патологическое дыхание Куссмауля), угнетением сознания, нарушением чувствительности, дисфункцией желудочно-кишечного тракта, резко выраженной дегидратацией тканей. Гиперкетонемия и кетонурия отсутствуют, могут выявляться незначительная гипергликемия и небольшая глюкозурия. Вследствие несвоевременной диагностики и трудности лечения прогноз может быть неблагоприятным.
4. Гипогликемическая кома.Связана с передозировкой инсулина, препаратов сульфонилмочевины, развитием вторичного гипопитуитаризма (следствие ангиопатии сосудов гипофиза), ослабляющего ответ на гипогликемию, и явлениями диабетического нефросклероза, что удлиняет время циркуляции инсулина и, кроме того, еще более снижает почечный порог для глюкозы, способствуя ее потере.
Причинами гипогликемии могут быть также гиперпродукция инсулина опухолью поджелудочной железы (инсулиномой), недостаточность контринсулярных гормонов, печеночные формы гликогенозов, заболевания печени, голодание, нарушение расщепления и всасывания углеводов в желудочно-кишечном тракте и др.
В механизме развития гипогликемической комы решающее значение имеет снижение доставки глюкозы к нервным клеткам, что ведет к их энергетическому истощению и нарушению функций ЦНС. При снижении уровня глюкозы менее 3 ммоль/л возникают потливость, тремор, чувство тревоги и голода, слабость. Затем развивается состояние, напоминающее алкогольное опьянение и сопровождающееся дезориентацией, агрессивностью, галлюцинациями. При дальнейшем падении содержания глюкозы (менее 2,5 ммоль/л) возникают клонические судороги и потеря сознания. В тяжелых случаях могут наступать отек и некроз отдельных участков мозга.
Микро- и макроангиопатииотносят к более поздним осложнениям, развивающимся при СД обоих типов.
1. Микроангиопатия.Это осложнение выражается в повреждении сосудов микроциркуляции и развивается вследствие метаболических нарушений в сосудистой стенке (гликозилирование белков) и развития васкулита. Чаще всего поражаются сосуды почек и сетчатки глаза.
Поражение почек - диабетическая нефропатияиз-за развития макро- и микроангиопатии в настоящее время является основной причиной ранней смертности у больных диабетом молодого возраста. При этом происходит избыточное гликозилирование коллагена базальных мембран почечных клубочков, приводящее к существенным нарушениям структуры и функций почечного фильтра.
Если в суточной моче концентрация альбумина превышает 30 мг (микроальбуминурия),и эти значения повторяются несколько раз, то необходимо проводить лечение, так как данные изменения характерны для начинающейся диабетической нефропатии. По мере прогрессирования поражения почек при диабете развивается выраженная протеинурия. Тщательный контроль за уровнем глюкозы в крови и лечение любых форм гипертонии могут приостановить микроальбуминуриюи предупредить развитие манифестной почечной недостаточности.
Поражение сетчатки глаз при диабете - диабетическая ретинопатияотносится к числу одной из наиболее частых причин развивающейся слепоты при этой патологии. Основную роль в патогенезе развития отечно-геморрагических форм диабетической ретинопатии играют максимальные суточные колебания уровня глюкозы в крови. Так, при повышении уровня глюкозы на 5,55
ммоль/л осмолярность сыворотки повышается на 5,5 мосмоль/л, что соответствует увеличению давления на 86 мм рт.ст., поэтому если увеличение концентрации сахара в крови идет быстро (в постабсорбтивном периоде), то в капиллярах сетчатки резко возрастает трансмуральное (разница между давлением внутри и снаружи сосуда) осмотическое давление. Жидкость «вытягивается» из ткани сетчатки в кровь, что ведет к резкому повышению трансмурального гидростатического давления в сосудах сетчатки. Повышение гидростатического давления за счет миогенной ауторегуляции приводит к сужению артериол вплоть до полного перекрытия кровотока (с развитием микроинфарктов сетчатки), а капилляры растягиваются с образованием микроаневризм и развития кровоизлияний.
Другой причиной, приводящей к потере зрения, является диабетическая катаракта,в патогенезе которой важную роль играет длительно существующая гипергликемия. Она вызывает, с одной стороны, усиление синтеза сорбитола и маннитола (ферментативное гликозилирование) и накопление этих углеводов в хрусталике глаза, приводящее по осмотическому градиенту к увеличению содержания в нем воды. С другой стороны, гликозилирование белков хрусталика и его капсулы (неферментативное) также вызывает необратимые нарушения его структуры.
2. Макроангиопатия.Диабетическая макроангиопатия проявляется атеросклерозом вследствие выраженного нарушения обмена липопротеинов. Особенностями атеросклероза при СД являются большая распространенность (захват многих сосудистых бассейнов), быстрое прогрессирование, начало в более молодом возрасте. Патологический процесс охватывает сосуды головного мозга, сердца, почек, а также сосуды конечностей, в особенности сосуды голени и стопы. Диабет, даже в условиях его лечения современными средствами, характеризуется ускоренными темпами старения организма. Наличием диабета обусловлены высокая частота инфарктов миокарда, инсультов и случаев гангрены пальцев ног или стопы.
В настоящее время считают, что диабетускоряет развитие атеросклерозав результате:
• дислипопротенемии(см. раздел 12.5.2);
• гиперхолестеринемии(см. раздел 12.5.8);
• эндотелиальной дисфункции,возникающей из-за окислительного стресса, основным фактором которого являются модифицированные ЛПНП;
• активации неспецифического звена системы иммунитета,сопровождающейся выработкой провоспалительных цитокинов;
• эффекта гормона роставследствие отсутствия противодействия со стороны инсулина в условиях его абсолютного или относительного дефицита, приводящего к усилению процесса пролиферации гладкомышечных клеток;
• усиленного синтеза тромбоксана А2.
Диабетическая нейропатия- нарушение функции нервов - развивается вследствие ангиопатии из-за нарушения кровоснабжения нервов, гликозилирования белков нейронов и миелиновых оболочек, гиперосмолярного поражения шванновских клеток (проявление гиперосмотической дегидратации при СД). Эти нарушения способны вызывать дисфункции любой системы организма, имитируя многочисленные неврологические заболевания. В патологический процесс могут быть вовлечены как чувствительные, так и двигательные или вегетативные нервные волокна. В качестве типичных примеров клинического проявления диабетических нейропатий можно назвать образование трофических язв на стопах - «диабетическую стопу», различные расстройства функций желудочно-кишечного тракта, мочевого пузыря, импотенцию. При исследовании структуры нервных волокон диабетиков с помощью световой микроскопии часто выявляются их демиелинизация, истончение и склероз эпиневрия, отек и дистрофии нервных волокон, глиальная клеточная реакция.
Патогенез диабетических нейропатий полностью не раскрыт, однако в настоящее время можно назвать ряд факторов, безусловно определяющих развитие этого осложнения диабета:
1) нарушение структуры миелина, когда патологический процесс обусловливает изменение как химической структуры, так и количественного соотношения между основными биохимическими компонентами (холестерин, ТАГ, фосфолипиды, гликолипиды и белки) миелина нервных волокон. Клинические наблюдения свидетельствуют о том, что под влиянием заместительной терапии инсулином происходит существенная коррекция многих из указанных сдвигов;
2) сорбитоловый путь окисления глюкозы - в результате данного процесса происходит ферментативное окисление глюкозы с образованием сначала сорбитола, а затем фруктозы, повышенное количество которых приводит к гиперосмотической дегидратации нейронов;
3) токсическое действие пирувата.
Следует отметить, что общая черта всех вышеперечисленных нарушений, по крайней мере до развития глубоких морфологических изменений, - их обратимость под влиянием терапии, приводящей к снижению гипергликемии.
Дата добавления: 2015-01-29; просмотров: 2053;