Типы объемных взаимодействий в белковых макромолекулах.
Водородные связи: силы Ван-дер-Ваальса; электростатические взаимодействия; поворотная изомерия и энергия внутреннего вращения. Общая конформационная энергия биополимеров
Типы объемных взаимодействий. Первичная структура полимерной цепи определяется химическими или валентными взаимодействиями. Объемные взаимодействия в основном определяют вторичную структуру макромолекул. Общим критерием стабильности молекулярной структуры является наличие минимума на кривой U(r) зависимости энергии взаимодействия от расстояния между взаимодействующими частями. Картинка «Электронный терм для двухатомной молекулы». 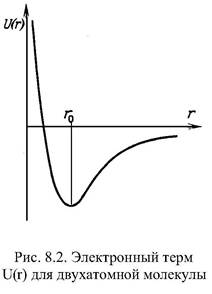
На малых расстояниях преобладают силы отталкивания, а на больших расстояниях превалирует притяжение. При r = ro силы притяжения и отталкивания уравновешивают друг друга. Значение энергии U(r) свободных частиц при r -> к бесконечности равно нулю, а энергия образованной ими стабильной структуры отрицательна U(ro) < 0. На малых расстояниях, где частицы отталкиваются, U(r) > 0. Минимум U(r0) соответствует максимальной по абсолютной величине и отрицательной по знаку энергии взаимодействия. В образовании вторичной структуры белка играют большую роль силы Ван-дер-Ваальса. Они имеют электромагнитную природу и связаны с взаимодействием электрических диполей в соседних молекулах. Наиболее распространены дисперсионные взаимодействия между молекулами, которые не обладают постоянными дипольными моментами. Природа этих сил носит квантовомеханический характер.
Неопределенности в значениях координаты дельта х и импульса дельта р связаны соотношением неопределенностей
дельта х дельта р = h
Это значит, что и в основном невозбужденном состоянии существуют быстрые смещения заряда электрона от положения равновесия, а следовательно, в молекуле в состоянии покоя появляются "мгновенные" дипольные моменты. Появление такого момента в одной молекуле индуцирует появление его в соседней молекуле. Возникает взаимодействие двух быстроменяющихся дипольных моментов, которые, таким образом, становятся связанными и притягиваются друг к другу. Энергия притяжения двух мгновенных диполей, или энергия дисперсионного взаимодействия, быстро убывает с расстоянием Uдисп~1/r6.
Кроме дисперсионного взаимодействия возможно и электростатическое притяжение между постоянными диполями в полярных молекулах. Кроме того, существуют также индукционные взаимодействия, которые возникают между постоянным дипольным моментом в одной молекуле и наведенным им диполем в соседней поляризуемой молекуле. Суммарное ван-дерваальсово взаимодействие двух молекул зависит от вклада всех типов дипольных взаимодействий и составляет по величине от 1,0 до нескольких десятков ккал/моль.
В выражении для полной энергии или полного потенциала взаимодействия необходимо учесть не только притяжение Uпритяж(r)~1/r6, но и отталкивание на близких расстояниях Uотт(r)~1/r12) сложение этих величин дает формулу на картинке 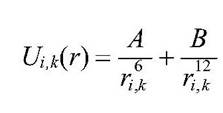 ,
,
где В - константы притяжения и отталкивания, r,k - расстояние между взаимодействующими атомами (i и k).
Наряду с силами Ван-дер-Ваальса большую роль в стабилизации биоструктур играют водородные связи и электростатические взаимодействия между заряженными и полярными группами. Водородные связи, например, стабилизируют вторичную структуру полипептидных цепей. В энергию водородной связи дают вклад электростатические взаимодействия, притяжение и отталкивание, а также энергия делокализации электронов. Величины энергии водородной связи сильно варьируют (3 - 8 ккал/моль). Так, водородная связь
О-Н ... О обладает энергией 8,6 ккал/моль.
Электростатические взаимодействия задаются формулой 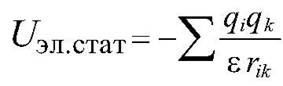 ,
,
где qi, qk - заряды на атомах (i и k), rik - расстояние между атомами, эйпсилон - диэлектрическая постоянная.
Внутреннее вращение и поворотная изомерия. Энергия ближних взаимодействий атомных групп зависит от расстояний между ними, которые в свою очередь меняются при вращении этих групп вокруг единичных связей. При близком расположении валентно не связанные атомы начинают отталкиваться, и возникает тормозящий энергетический потенциал, препятствующий вращению атомных групп. Энергия вращения атомных групп вокруг единичных связей дает основной вклад в общую конформационную энергию полимерной цепи.
Картинка «График зависимости потенциальной энергии»
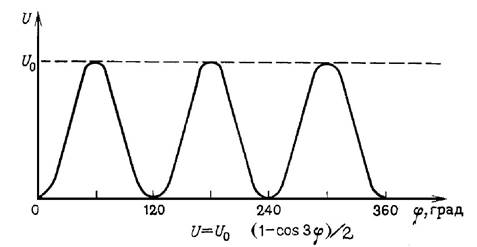
- молекула этана имеет минимум конформационной энергии в трансконформации и максимум - в цисконформации. Энергетический барьер, или тормозящий потенциал, для перехода одной трансконформации в другую через цис-форму при повороте вокруг С-С связи на 120° равен~3 ккал/моль. Зависимость потенциала внутреннего вращения от угла поворотафи задается выражением
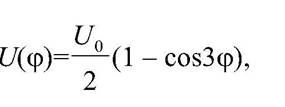
где U0 - высота барьера.
Общая конформационная энергия полимера зависит от взаимных углов поворотов звеньев вокруг единичных связей. Подобная система, где энергия составляющих элементов зависит от их взаимодействия друг с другом, называется кооперативной.
Конформационная энергия полипептидной цепи определяется всеми видами объемных взаимодействий и зависит от энергии внутреннего вращения боковых цепей аминокислотных остатков вокруг единичных связей. Общее строение полипептидной цепи представлено на картинке «строение ППЦ»
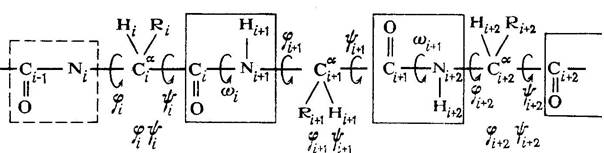
Двойной характер пептидной связи C альфа iO-Ni+1H препятствует врашению вокруг нее. Он обусловлен обобшествлением неподеленной пары 2 S2 электронов атома азота между азотом и углеродом. Вследствие этого происходят выталкивание электрона углерода из двойной пи - связи C=O и локализация его на кислороде с частичным преврашением связи C=O в одиночную. Картинка «связи».
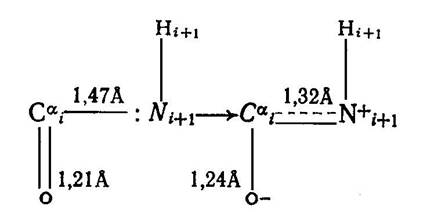
Делокализация электронов между атомами N, С, О приводит к тому, что пептидная группа максимально стабилизируется, когда ее атомы, включая альфа - углеродные атомы соседних аминокислот, расположены в одной плоскости. Поэтому врашение вокруг пептидной связи Cai=Ni+1 затруднено в силу ее двойного характера. Можно учитывать только врашение вокруг связей Ni - Cai (угол фi) и Cai - Ci (угол psii ), так как в такой цепи отсутствуют стерические перекрытия атомов i - й пептидной единицы с (i+2) - й или (i - 2) - й единицами. В пептидной цепи имеет место только попарное кооперативное взаимодействие при врашении вокруг единичных связей, принадлежаших одному и тому же aльфа - углеродному атому. Каждая пара углов (фиi и psii) может рассматриваться независимо, а кооперативность в цепи фактически ограничивается взаимодействием соседних пептидных единиц. Потенциалы внутреннего врашения Uо вокруг единичных связей весьма малы (~1,0 ккал/моль). Следовательно, минимумы отдельных дискретных состояний, возникающих при изменении углов фи и psi, разделены невысокими барьерами. Обшее выражение для конформационной энергии имеет вид картинка «конформационная энергия».
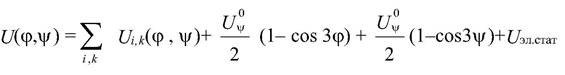
Конформация простейших фрагментов полипептидной цепи может быть найдена путем расчета. Для этого необходимо знать химическую последовательность аминокислотных остатков и подсчитать энергию невалентных взаимодействий их атомов и атомных групп по формуле. Найденная в результате конформация задается в виде конкретных значений углов поворота атомных групп и соответствующих расстояний между ними, при которых конформационная суммарная энергия, зависящая от всех видов объемных взаимодействий, достигает минимальных значений.
Дата добавления: 2016-04-11; просмотров: 3934;