Глава 8 Неингаляционные анестетики
Общая анестезия достигается не только с помощью ингаляционных анестетиков. Различные препараты, назначаемые внутрь, внутримышечно и внутривенно, вызывают анестезию или потенцируют ее. Седативные средства для премедикации, которым посвящен "Случай из практики", приведенный в настоящей главе, назначают внутрь или внутримышечно. У взрослых применяют внутривенную индукцию анестезии. Даже поддержание общей анестезии можно осуществить с помощью методики тотальной внутривенной анестезии (TBBA, см. "Случай из практики", гл. 46). Данная глава начинается с обзора принципов фармакоки-нетики и фармакодинамики в приложении к рассматриваемой группе препаратов. Затем проводится клиническая фармакология отдельных групп: барбитуратов, бензодиазепинов, опиоидов, кета-мина, этомидата, пропофола и дроперидола.
Фармакологические принципы
Фармакокинетика
Как уже было отмечено выше в гл. 7, фармакокине-тикой называется учение о взаимоотношениях между дозой, концентрацией в тканях и продолжительностью действия лекарственного средства. Иными словами, фармакокинетика описывает, что организм делает с препаратом. Фармакокинетика определяется четырьмя параметрами: абсорбцией, распределением, биотрансформацией и экскрецией. Элиминациявключает удаление препарата путем биотрансформации и экскреции. Клиренс— мера скорости элиминации.
Абсорбция
Лекарственное средство может попасть в системный кровоток несколькими путями: при назначении внутрь, сублингвально, ректально, через легкие, чрескожно, подкожно, внутримышечно и внутривенно. Абсорбция — это процесс, в ходе которого лекарственный препарат из места введения поступает в системный кровоток. На абсорбцию влияют физические свойства препарата (растворимость, рКа и концентрация) и характеристики места абсорбции (перфузия, рН и площадь поверхности). Следует отличать абсорбцию от биодоступности,которая представляет собой фракцию неизмененного вещества в плазме крови относительно исходной дозы препарата. Например, нитроглицерин хорошо абсорбируется через ЖКТ, но при приеме внутрь имеет низкую биодоступность, потому что подвергается интенсивному метаболизму в печени (так называемый эффект первого прохождения).
Назначение препарата внутрьудобно, экономично и позволяет достаточно точно его дозировать. Тем не менее на поступление препарата в системный кровоток влияют возможность контакта с больным, эффект первого прохождения, рН желудка, секреторная и моторная функции ЖКТ, пища, другие лекарственные средства.
Абсорбируется преимущественно неионизированная фракция препарата. Следовательно, препараты-кислоты лучше всасываются в кислой среде (Кис- + H+ → КисН), препараты-основания — в щелочной (ЩН+ → H+ + Щ).
Кровь из сосудов полости рта дренируется непосредственно в верхнюю полую вену, поэтому при сублингвальном и буккальномпутях введения препараты поступают в системный кровоток, минуя печень. Ректальныйпуть — альтернатива приему внутрь при невозможности контакта с пациентом (например, у детей) или при физической невозможности такого приема. Венозная кровь из прямой кишки поступает в нижнюю полую вену, минуя печень, поэтому при ректальном пути введения биодоступность выше, чем при приеме внутрь. При ректальном введении нельзя быть уверенным в точности дозировки; кроме того, многие препараты раздражают слизистую оболочку прямой кишки. Абсорбция ингаляционныханестетиков обсуждается в гл. 7.
К преимуществам чрескожноговведения относятся длительная непрерывная абсорбция, возможность использования незначительных доз препарата. Роговой слой служит эффективной преградой для большинства соединений, за исключением низкомолекулярных жирорастворимых препаратов (например, клонидин, нитроглицерин, скополамин).
Наконец, препараты вводят парентерально, т. е. подкожно (п/к), внутримышечно (в/м) и внутривенно (в/в).Абсорбция препарата при подкожном и внутримышечном введении определяется диффузией из места инъекции в кровь. Скорость диффузии зависит от местного кровотока и среды-переносчика (растворы абсорбируются быстрее, чем суспензии). Некоторые препараты могут вызывать боль при введении и некроз тканей. При внутривенной инъекции препарат полностью поступает в системный кровоток.
Распределение
Распределение — ключевой параметр фармакоки-нетики, определяющий концентрацию препарата в органе-мишени. Распределение лекарственного средства зависит от перфузии органа, связывания препарата с белками и его жирорастворимости.
После абсорбции лекарственное средство поступает в системный кровоток. Органы с высокой перфузией (группа хорошо васкуляризованных тканей)поглощают диспропорционально большее количество лекарственных средств, чем органы с низкой перфузией (мышцы, жир и слабо васкуля-ризованные ткани).Поэтому, несмотря на относительно небольшую массу, хорошо васкуляризован-ные ткани поглощают значительное количество препарата (табл. 8-1).
Пока лекарственное средство связано с белком плазмы, оно не доступно для поглощения органом вне зависимости от интенсивности кровотока. Альбумин связывает главным образом препараты-кислоты (например, барбитураты), в то время как α1-гликопротеин — препараты-основания (например, местные анестетики). Если концентрация белков плазмы снижена или места связывания на них заняты (например, другими лекарственными средствами), то количество доступного для поглощения тканью свободного препарата увеличивается. Болезни печени и почек, хроническая сердечная недостаточность и злокачественные новообразования снижают выработку альбумина. Травма (включая хирургическую операцию), инфекции, инфаркт миокарда и хронические болевые синдромы — все это увеличивает выработку α1-гликопротеина.
Доступность препарата для органа еще не гарантирует поглощения его этим органом. Например, поступление ионизированных препаратов в ЦНС резко ограничено гематоэнцефалическим барьером,который образован плотными контактами перикапиллярных глиальных клеток и эндоте-лиальных клеток. Жирорастворимые неионизированные молекулы свободно проходят через липидные мембраны. Другие факторы, такие как размер молекулы и поглощение препарата в легких, также влияют на распределение.
После того как в ходе начального распределения насыщаются хорошо васкуляризованные ткани, большая масса слабо васкуляризованных тканей продолжает поглощать препарат из кровотока. Когда концентрация препарата в плазме значительно снижается, некоторое количество его покидает хорошо васкуляризованные ткани и поступает в кровоток, чтобы поддержать равновесие. Это перераспределениеиз хорошо васкуляризованных тканей приводит к прекращению действия многих анесте-тиков. Например, пробуждение после анестезии ти-опенталом обусловлено не метаболизмом или экскрецией, а перераспределением препарата из головного мозга в мышцы. Отсюда следует логический вывод: если насытить препаратом слабо васкуляризованные ткани (например, с помощью повторных введений), то перераспределения не будет и окончание эффекта препарата станет определяться элиминацией. Поэтому препараты короткого действия, такие как фентанил и тиопентал, после повторных введений или после введения большой однократной дозы действуют значительно дольше.
Кажущийся объем, в котором распределено лекарственное средство, называется объемом распределения (Vd,от англ, volume of distribution). Объем распределения равен частному от деления дозы препарата на концентрацию в плазме:
Vd = Доза/Концентрация.
ТАБЛИЦА 8-1. Группы тканей: состав, доля массы тела, доля сердечного выброса
| Группа тканей | Состав | Доля массы тела, % | Доля сердечного выброса, % |
| Хорошо васкуляризованные | Мозг, сердце, печень, почки, эндокринные железы | ||
| Мышцы | Мышцы, кожа | ||
| Жир | Жир | ||
| Слабо васкуляризованные | Кости, связки, хрящ | О |
Вычисления осложняются необходимостью учитывать влияние элиминации и перераспределения. Низкий объем распределения указывает на то, что препарат распределяется главным образом в кровь (при массе 70 кг Vd панкурония равен 10 л). К причинам низкого объема распределения относят высокую степень ионизации или связывания с белками. В то же время объем распределения может превышать объем общей воды организма (приблизительно 40 л). Это объясняется высокой растворимостью или лучшим связыванием препарата в тканях по сравнению с плазмой (например, Vd фентанила равен 350 л). Итак, объем распределения не представляет собой реальный объем, а отражает объем плазмы, который был бы необходим для распределения дозы препарата в измеренной концентрации.
Биотрансформация
Биотрансформация — это химическое превращение лекарственного вещества в ходе метаболизма. Конечные продукты метаболизма обычно (но не всегда) неактивные и водорастворимые. Последнее свойство обеспечивает экскрецию через почки. Главным органом биотрансформации является печень.
Метаболическую биотрансформацию подразделяют на реакции I и II фазы. Реакции I фазыпреставляют собой окисление, восстановление или гидролиз, в ходе которых молекула лекарственного средства становится более полярной. В реакциях II фазы,или реакциях конъюгации, к молекуле лекарственного средства (или его метаболита, образовавшегося в результате реакции I фазы) присоединяется молекула эндогенного вещества, например глюкуроновой кислоты, в результате чего образуется более полярный метаболит, легко выводимый с мочой. В подавляющем большинстве случаев конъюгация следует за реакцией I фазы, но иногда метаболическая трансформация ограничивается исключительно реакцией I фазы или же конъюгация предшествует реакции I фазы.
Печеночный клиренс— это скорость элиминации лекарственного вещества в результате его биотрансформации в печени. Точнее, клиренс — это объем плазмы, очищенный от препарата за единицу времени; клиренс измеряют в мл/мин. Печеночный клиренс зависит от печеночного кровотока и фракции препарата, поглощаемого из крови печенью (отношение печеночной экстракции).С одной стороны, препараты, которые поглощаются печенью в значительной степени, имеют высокое отношение печеночной экстракции и их клиренс пропорционален печеночному кровотоку. С другой стороны, препараты с низким отношением печеночной экстракции поглощаются печенью незначительно и их клиренс ограничен емкостью фермент-них систем печени. При болезнях печени на фармакокинетику лекарственного средства влияет не только отношение печеночной экстракции, но также степень снижения печеночного кровотока и дисфункции гепатоцитов.
Экскреция
Главным органом экскреции являются почки. Лекарственные вещества, не связанные с белками плазмы, свободно проходят через клубочковый фильтр. Неионизированная фракция препарата реабсорбируется в почечных канальцах, а ионизированная — выделяется с мочой. Таким образом, изменение рН мочи влияет на почечную экскрецию. Почечный клиренс— это скорость элиминации препарата путем почечной экскреции. Почечная недостаточность влияет на фармакокинетику многих лекарственных средств, изменяя степень связывания с белками, объем распределения и почечный клиренс.
Исключительно от экскреции с желчью зависит элиминация относительно небольшого количества лекарственных средств, потому что в кишечнике эти средства большей частью реабсорбируются и в конце концов выводятся с мочой. Отсроченные токсические реакции некоторых препаратов (например, фентанила) объясняются именно энтеро-гепатической рециркуляцией.
Элиминация ингаляционных анестетиков обеспечивается легкими (гл. 7).
Модели камер
Модели камер представляют собой упрощенную схему, позволяющую охарактеризовать распределение и элиминацию лекарственных средств в организме. Камерой называют группу тканей, обладающих сходными фармакокинетическими характеристиками. Например, плазма и хорошо вас-куляризованные ткани — это центральная камера,в то время как мышцы, жир и кожа — периферическая камера.Следует иметь в виду, что под камерами понимают воображаемые пространства, а не реальные анатомические структуры.
Двухкамерная модельхорошо коррелирует с распределением и элиминацией многих лекарственных средств (рис. 8-1). После в/в струйного введения концентрация препарата в плазме мгновенно возрастает. Начальное быстрое снижение концентрации препарата в плазме, называемое фазой распределения,или альфа(α)-фазой,соответствует перераспределению препарата из центральной камеры в периферическую. После того как распределение замедляется, элиминация из центральной камеры вызывает длительное, но менее крутое снижение концентрации препарата в плазме, что носит название фазы элиминации,или бета(β)-фазы.Период полусуществования препарата в фазе элиминации прямо пропорционален объему распределения и обратно пропорционален клиренсу. Кривые концентрации многих лекарственных средств лучше описывать с помощью трехкамер-ной модели,где оперируют одной центральной камерой и двумя периферическими. Концентрация лекарственного вещества в плазме после в/в струйного введения описывается следующим трех-экспоненциальным уравнением:
Cp(t) = Ae-at + Be-βt + Ce-γt
где Cp(t) — концентрация препарата в плазме в момент времени t; A, В и С — фракционные коэффициенты, которые указывают на относительный вклад каждой из трех констант периодов полусуществования препарата (а соответствует периоду полусуществования в фазе быстрого распределения, β — в фазе медленного распределения, γ — в фазе окончательной элиминации). Следовательно, концентрация препарата в плазме определяется шестью фармако-кинетическими параметрами, и не все из них являются периодами полусуществования, как часто ошибочно считают.
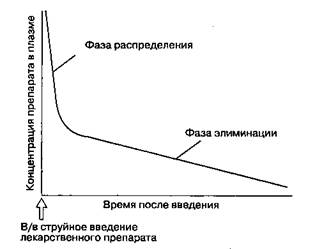
Рис. 8-1. Двухкамерная модель описывает фазу распределения (α-фазу) и фазу элиминации (β-фазу). Во время фазы распределения лекарственный препарат поступает из центральной камеры в периферическую. Фаза элиминации состоит в метаболизме и экскреции препарата
Фракционные коэффициенты так же важны для расчета продолжительности действия препарата, как периоды полусуществования. Например, периоды полусуществования в фазах распределения и элиминации для лекарственного средства х могут быть больше по сравнению с таковыми препарата у, но концентрация препарата х в плазме может снижаться значительно быстрее только вследствие того, что его фракционный коэффициент распределения (А) больше. Иными словами, если распределение, а не элиминация играет основную роль в снижении концентрации препарата, то даже при длительных периодах полусуществования концентрация препарата в сыворотке будет быстро уменьшаться. Следовательно, продолжительность действия препарата нельзя рассчитать, зная только периоды полу существования.
Скорость распределения и биотрансформации принято описывать в терминах кинетики первого порядка.Другими словами, в единицу времени распределяется или подвергается метаболизму постоянная фракция (доля) препарата вне зависимости от концентрации препарата в плазме. Например, каждый час подвергается биотрансформации 10 % препарата вне зависимости от того, будет ли его концентрация в плазме равна 10 мкг/мл или 100 мкг/мл. Если концентрация препарата превышает возможности биотрансформации, то в единицу времени подвергается метаболизму одинаковое количество препарата (кинетика нулевого порядка).Можно привести аналогичный первому пример: каждый час будет подвергаться метаболизму 500 мкг препарата вне зависимости от того, будет ли его концентрация в плазме равна 10 мкг/мл или 100 мкг/мл. Метаболизм алкоголя описывается кинетикой нулевого порядка.
Фармакодинамика
Фармакодинамика — это наука о действии препарата на организм, включая токсические реакции (т. е. о том, как лекарственный препарат влияет на организм). Действие препарата на организм характеризуется эффективностью, мощностью и терапевтической широтой. Фармакодинамика также изучает механизмы действия, соотношение между структурой и активностью и межлекарственные взаимодействия. Изучение рецепторов лекарственных средств и графиков "доза-эффект" облегчает понимание фармакодинамики.
Кривые "доза-эффект"
Кривые "доза-эффект" отражают зависимость между дозой препарата и фармакологическим эффектом. Дозу препарата (или его устойчивую концентрацию в плазме) откладывают по оси абсцисс (т. е. по оси х) в линейном (рис. 8-2А) или логарифмическом (рис. 8-2Б) выражении. Величину фармакологического эффекта откладывают по оси ординат (т. е. по оси у) в виде абсолютных единиц (см. рис. 8-2A) или доли максимального эффекта (см. рис. 8-2Б). Крутизна наклона кривой свидетельствует о мощностипрепарата. Эффективность определяется максимальным эффектом препарата. Наклон кривой отражает связывание препарата с рецептором. Изучив зависимость фармакологического эффекта от концентрации препарата в сыворотке, можно нейтрализовать влияние фармако-кинетики на кривые "доза-эффект"
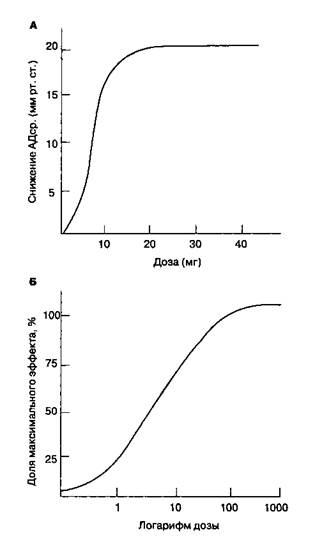
Рис. 8-2. Форма кривой "доза-эффект" зависит от линейного (А) или логарифмического (Б) отображения дозы препарата или его устойчивой концентрации в сыворотке
Медиана эффективной дозы(ЭД50) — это доза препарата, которая вызывает ожидаемый эффект у 50 % популяции. Следует отметить, что ЭД50 не является дозой, способной вызвать половину максимального эффекта. Для ингаляционных анестетиков ЭД50 соответствует минимальной альвеолярной концентрации (1 МАК, см. гл. 7). Медиана летальной дозы(ЛД50) — это доза препарата, которая приводит к смерти 50 % популяции. Терапевтическим индексом называют отношение медианы летальной дозы к медиане эффективной дозы (ЛД50 : ЭД50).
Дата добавления: 2016-03-30; просмотров: 1165;