Биохимические и биологические методы анализа
Спектральный анализ – основан на изучении спектров излучения различных веществ. Пробы анализируемого вещества «сжигают» в определенных условиях, вещество испаряется диссоциирует на атомы, которые возбуждаясь дают спектр. Излучаемый при этом свет, проходя через стеклянную призму спектроскопа, разлагается на свои составные части (разные света) и экспериментатор наблюдает ряд различных линий (линейный спектр). По линиям судят о присутствии того или иного элемента в анализируемом продукте.
Чем выше интенсивность линий, тем выше концентрация вещества. С помощью спектрографов можно сфотографировать излучение и по степени почернения линий на фотопластинке определить концентрацию вещества метод высокочувствителен, примеси веществ определяет до 0,0001% - десятичных долей процента. Метод применяется при определение минеральных состава продуктов растительного и животного происхождения.
Люминесценция – свечение атомов, ионов, молекул и более сложных частиц вещества, которое возникает в результате перехода в них электронов при возвращении из возбужденного состояния в нормальное. Для перевода частиц в возбужденное состояние подводят определение количество энергии. Свечение или часть энергии выделяется в виде квантов люминесценции. Этот метод используется для определения витаминов, белков и жиров в молоке, для определения свежести мяса и рыбы и различной порчи овощей, плодов для обнаружения в продуктах питания консервантов, лекарственных препаратов, канцерогенных веществ, пестицидов.
Например: здоровый картофель на разрезе имеет желтую флуоресценцию (т.е. собственное свечение). При поражении картофеля фитофтора - становиться интенсивно голубой, при поражении кальциевой гнилью – зеленоватой, при появлении вирусных заболеваний – разного цвета, преимущественно в сосудистой части клубня.
Лимоны и апельсины имеют флуоресценцию с голубоватым оттенком, маринады – темно-оранжевую с фиолетовым оттенком. При поражении голубой плесенью появляется темно-синяя флуоресценция в виде пятен в местах поражения.
Таким образом, изменение флуоресценции свежих плодов и овощей позволяет обнаружить на очень ранней стадии начало порчи, что необходимо при хранение, длительной транспортировке и консервировании. И в молоке и в мясе, жирах и масле можно с помощью этого метода обнаружить порчу или примеси других продуктов т.е. установить чистоту продукта, идентифицировать его, а также вид мяса (говядина, или свинина).
При оценке качества пищевых продуктов большое значение уделяется их консистенции. Существуют реологические методы оценки консистенции – первичной оценки пищевых продуктов.
Реология изучает структурно-механические свойства материалов (деформацию). К реологическим свойствам относятся вязкость, упругость, эластичность и прочность.
Вязкость – свойство газов, жидкостей и твердых тел сопротивляться действию внешних сил (т.е. перемещению слоев). Для твердых тел (корнеплодов) – сопротивляться развитию деформации.
Упругость – способность тел сопротивляться изменению их объема формы под действием внешних сил или по другому – способность тела восстанавливать свою форму после снятия нагрузки (затяжное тесно – где меньше сахара и жира). Эластичность – способность материала при незначительных усилиях восстанавливаться без разрушений, т.е. упруго - вязкими и т.д. все перечисленные свойства проявляются при обработке этих материалов или сырья, являются полезными или наоборот мешают, создают дополнительные трудности.
Например: при уменьшении количества клейковины в муке вы будете наблюдать проявление этих реологических свойств (упругость, эластичность и т.д.) на знание этих свойств созданы соответствующие аппараты для производства конфет (образование корпусов конфет), замес теста, штампование макаронных изделий и т.д.
Возможно, появление новых более совершенных методов, которые приобретут массовый характер, т.е. каждый потребитель будет иметь возможность при покупке определить качество продукта при помощи мини прибора (таких как измеритель радиоактивного фона и т.д.).
Основная цель данной лекции в том, что при производстве или исследовании продуктов питания вы тщательно подбирали самый эффективный метод и способствовали тому, чтобы потребитель получал качественный ценный продукт питания.
Молекулярные сита, сорбенты, избирательно поглощающие из окружающей среды вещества, молекулы которых не превышают определённых размеров. Такие сорбенты как бы отсеивают крупные молекулы от мелких. Различают минеральные (неорганические) и органические М. с. Неорганические М. с. имеют жёсткую кристаллическую структуру, в которой находятся полости, соединённые между собой узкими каналами «порами» или «окнами». Малые размеры «окон» препятствуют диффузии крупных молекул во внутренние полости сорбента. Некоторые алюмосиликаты — природные и синтетические цеолиты — характерные представители М. с. этого типа.
Флуоресценция - люминесценция, характеризуемая небольшим временем свечения после прекращения возбуждения.
Светопоглощение ( оптическую плотность, абсорбцию) Вычисляют по формуле:
A=lg I0/It
I0 – интенсивность входящего светового потока.
It – интенсивность прошедшего светового потока.
Оптическая плотность зависит от толщины светопоглощающего слоя, от концентрации растворённого светопоглощающего вещества:
A=lg I0/It=k1*l – закон Бугера-Ламберта.
k1 – коэффициент пропорциональности;
l – толщина светового слоя, см.
Оптическая плотность зависит и от концентрации растворённого светового вещества:
A=lg I0/It=k11*l – закон Бугера-Ламберта.
k11 – коэффициент пропорциональности;
l – толщина светового слоя, см.
Закон Бера справедлив, если при изменении концентрации вещества не происходит его диссоциация, гидролиз комплексообразование и другие реакции.
В фотометрических методах анализа применяют объеденённый заклн бугера-Ламберта-Бера:
A=lg I0/It=k*l*с
При концентрации раствора выраженной в моль на литр и длинна в см коэффициент пропускания К называют молярным коэффициентом светопоглощения e.
Физический смысл e: оптическая плотность 1 моль/литр раствора измеренная в кювете длинной 1 см.
Закон Бугера-Ламберта-Бера: оптическая плотность раствора прямопропорцианальна концентрации светопоглощающего вещества, толщине слоя раствора и молярному коэффициенту светопоглощения.
Аналитическим сигналом в кинетических методах является скорость процесса или пропорциональная ей величина.
Реакцию, скорость которой измеряется, называют индикаторной, а вещество, по изменению концентрации которого судят о скорости процесса — индикаторным веществом.
Индикаторные реакции могут быть основаны на катализе окислительно-восстановительных реакций, реакций замещения в координационной сфере ионов металлов, реакций гидролиза и декарбоксилирования органических соединений. Наиболее чувствительны и сравнительно просто выполнимы окислительно-восстановительные каталитические реакции. Они чаще всего используются в анализе неорганических веществ.
Кроме каталитических реакций в кинетических методах используют и некаталитические реакции окисления-восстановления, разложения, осаждения.
К индикаторной реакции предъявляют ряд требований:
· концентрация определяемого компонента за время наблюдения практически не должна меняться. Катализатор в ходе реакции не расходуется. Если же определяемым является одно из реагирующих веществ (некаталитический вариант метода), то с достаточной точностью его можно определять в тот начальный период, когда его концентрация изменяется не более чем на 5%;
· необходимо наличие быстрого, простого и доступного метода наблюдения за скоростью индикаторной реакции, т. е. за изменением концентрации индикаторного вещества во времени.
· скорость индикаторной реакции должна находиться в определенных пределах. Оптимальное время наблюдения за скоростью индикаторной реакции 5–15 мин. Однако с развитием методов изучения быстрых процессов все чаще используют реакции, протекающие с большой скоростью.
Существуют два варианта кинетических методов:
В каталитическом варианте кинетического метода (каталитическом методе, каталиметрии) определяемый компонент или связанные с ним соединения являются катализатором индикаторной реакции.
В некаталитическом варианте кинетического метода определяемым компонентом является одно из реагирующих веществ в некаталитической или каталитической индикаторной реакции.
Каталитические методы отличаются от других химических методов анализа высокой чувствительностью, а некаталитический вариант кинетических методов – высокой селективностью.
Если концентрация хотя бы одного из реагирующих веществ за время наблюдения за скоростью реакции заметно меняется (более чем на 10%), то между концентрацией индикаторного вещества и временем существует более сложная (например, логарифмическая, обратная и т. д.) зависимость. Такой кинетический метод называют интегральным. В интегральном варианте часто применяют построение зависимостей концентрации индикаторного вещества от времени в полулогарифмических, обратных или других координатах, т. к. для расчета неизвестной концентрации определяемого компонента удобнее использовать прямоугольные участки кинетических кривых. Характер кинетических кривых, а следовательно, и использование дифференциального или интегрального вариантов кинетических методов анализа определяется типом индикаторной реакции, ее механизмом.
В настоящее время наиболее распространенными являются три основных способа определения неизвестной концентрации по данным кинетических измерений. Это способы тангенсов, фиксированного времени, фиксированной концентрации. Рассмотрим их применительно к дифференциальному варианту кинетического метода анализа.
Способ фиксированного времени. При определенном, строго фиксированном интервале времени протекания реакции, измеряют концентрацию индикаторного вещества в пробах с известными концентрациями определяемого компонента. Градуировочный график строят в координатах концентрация определяемого вещества — концентрация индикаторного вещества при фиксированном времени протекания реакции tфикс. Часто при работе этим методом индикаторную реакцию останавливают при tфикс.. Путем резкого охлаждения, изменения кислотности раствора, добавления ингибиторов.
Способ фиксированной концентрации. В отдельных пробах с известными концентрациями определяемого вещества проводят индикаторную реакцию до строго определенной (фиксированной) концентрации индикаторного вещества хфикс. и измеряют время достижения этой концентрации. Градуировочный график строят в координатах: концентрация определяемого компонента — величина, обратная времени достижения хфикс..
В интегральном варианте все способы определения неизвестной концентрации аналогичны, лишь между временем реакции и концентрацией индикаторного вещества существует более сложная функциональная зависимость. В этом случае находят функции концентрации индикаторного вещества, линейно изменяющиеся во времени (логарифмическая, обратная и т. д.).
Каталиметрическое титрование — процесс титрования в присутствии катализатора, в котором точку конца титрования определяют по резкому увеличению или уменьшению скорости реакции.
С целью автоматизации каталиметрического метода анализа скорость реакции часто измеряют в открытых системах. Открытой называют систему, в которую по мере протекания реакции вводят реагенты и/или из которой отводят продукты реакции. В ходе реакции растворы подаются в реакционную камеру с постоянной или регулируемой скоростью. Разработаны разные варианты открытых систем: на основе проточных методов и «стат»-методов.
Проточные методы. К ним относится метод непрерывной струи, основанный на смешении реагентов в струе и предложенный для быстро протекающих реакций с периодом полупревращения t1/2 = 0,01–10 с. Другой вариант проточного метода применяют для измерения скоростей сравнительно медленно протекающих реакций с t1/2 = 1–10 мин. В этом случае проточная ячейка одновременно является и смесительной камерой. Исходные реагенты индикаторной реакции и анализируемый раствор, содержащий катализатор с концентрацией скат, непрерывно подаются насосами в смесительную камеру вместимостью около 10 мл, продукты реакции и реагенты вытекают со скоростью 2–20 мл/мин. При каждом значении скат устанавливается постоянная концентрация индикаторного вещества и фиксируется постоянный сигнал, соответствующий скат. Смена раствора в кювете происходит за 1–2 мин, что определяет производительность анализатора 30 проб в час.
Стат-метод предполагает введение реагентов со скоростью, равной скорости их расходования в реакции, так что концентрация индикаторного вещества остается постоянной. Скорость введения реагента регулируется автоматически.
Воспроизводимость результатов кинетических измерений повышается при использовании метода одновременного компарирования. В анализируемый раствор и растворы шкалы стандартов одновременно с помощью стартовой пипетки вводят реагент, инициирующий протекание каталитической реакции. Через определенный промежуток времени сравнивают аналитические сигналы анализируемого раствора и шкалы стандартов и оценивают содержание определяемого вещества. Метод не требует термостатирования.
Для учета влияния примесей на скорость реакции применяют метод добавок. Скорость реакции измеряют в равных аликвотных частях анализируемого раствора без добавки и в присутствии определенных добавок катализатора. Метод добавок дает правильные результаты, если в растворе отсутствуют посторонние примеси, обладающие каталитическим действием на индикаторную реакцию.
Скорость реакции можно определять по времени внезапного появления окраски раствора в реакциях Ландольта. Реакции Ландольта — это медленные химические реакции, в которых образование окрашенного продукта реакции задерживается подходящим реагентом, специально добавленным для этой цели. Например, при окислении бромида персульфатом, катализируемом ионами меди(II), образующийся бром окисляет аскорбиновую кислоту и не взаимодействует с индикатором N,N-диметил-п-фенилендиамином. Когда практически вся аскорбиновая кислота окислится, появляется окраска индикатора. Метод, основанный на эффекте Ландольта, в ряде случаев обеспечивает более высокую воспроизводимость результатов анализа, чем обычный метод фиксированной концентрации, разновидностью которого он является.
Концентрацию катализатора можно определять по длительности индукционного периода tинд., по истечении которого скорость реакции становится заметной (рис. 2). Этот способ является разновидностью метода фиксированной концентрации. Индукционный период наблюдается не только в реакциях Ландольта, но и в автокаталитических реакциях, а также в реакциях, когда в начальный период изменяется соотношение форм катализатора. Длительность индукционного периода связана с концентрацией катализатора зависимостью
За изменением концентрации индикаторного вещества во времени можно наблюдать любым методом, и при построении кинетических кривых вместо концентрации образующегося продукта использовать любую, пропорциональную ей величину — оптическую плотность, силу тока, потенциал системы и т. д. Чаще всего для наблюдения за скоростью индикаторной реакции используют спектрофотометрические и люминесцентные, реже — электрохимические, термометрические и титриметрические методы.
Некаталитические методы не отличаются высокой чувствительностью (она определяется, как правило, методом наблюдения за скоростью индикаторного процесса), но они селективны, часто позволяют определять в смеси близкие по свойствам вещества без их предварительного разделения. Эти методы применяют при анализе смесей органических соединений (спиртов, сахаров, аминов) и смесей таких близких по свойствам ионов металлов, как щелочно-земельные и редкоземельные элементы.
Каталитические методы анализа отличаются высокой чувствительностью, которая для многих неорганических веществ сравнима с чувствительностью масс-спектральных и активационных методов анализа, а для органических — с наиболее чувствительными вариантами хроматографии. В отдельных случаях, например, для серебра, хрома, кобальта, каталитические методы — наиболее чувствительные из всех известных методов анализа. При этом преимуществом каталитических методов является сочетание высокой чувствительности с простотой аппаратурного оформления и методики проведения анализа.
Среди каталитических методов высокую чувствительность и селективность имеют ферментативные методы, основанные на использовании реакций, катализируемых ферментами. Ферментативными методами определяют субстраты, сами ферменты и эффекторы ферментов (соединения, мешающие активности ферментов). Методы определения субстратов — веществ, на которые действуют ферменты — высокоселективны и даже специфичны, что позволяет определять субстраты непосредственно в матрице сложных объектов (кровь, биомассы и биожидкости, многокомпонентные технологические растворы). Чувствительность определения при этом обусловлена методом, выбранным для контроля за скоростью процессов. Часто в этих случаях используют ферментные электроды. Методы определения эффекторов ферментов высокочувствительны, но не всегда селективны.
В кинетических методах наиболее часто используют метод тангенсов как наиболее точный (использует большое число экспериментальных данных) и универсальный (применим для реакций с индукционным периодом). Реже применяют способ фиксированного времени и способ фиксированной концентрации, хотя эти способы более просты и менее трудоемки. Способ фиксированной концентрации используют обычно при автоматизации контроля, способ фиксированного времени — при проведении серийных анализов.
Метод каталиметрического титрования применяют для определения с повышенной точностью микросодержания ионов металлов или органических соединений, образующих с ионом металла устойчивые, каталитически неактивные комплексы, и следов органических веществ в неорганических солях особой чистоты. При титровании органического соединения избыток титранта (иона металла-катализатора) уже в концентрации 10–8–10–6 М вызовет протекание каталитической реакции и тем самым определит конечную точку титрования .
Основное применение каталитических методов в анализе реактивов и веществ особой чистоты — определение микроконцентраций переходных металлов. Именно каталитические методы позволяют определить ионы элементов, содержание которых часто лимитируется в технических условиях на вещества особой чистоты табл. 1.
Чистота цитирования примесей* в веществах особой чистоты [16] и пределы обнаружения их каталиметрическими методами
| Примесь | Чистота цитирования, % | Предел обнаружения, нг/мл | Примесь | Чистота цитирования, % | Предел обнаружения**, нг/мл |
| Fe | Ag | ||||
| Cu | Pb | ||||
| Ni | Ti | ||||
| Mn | 0,1 | V | 0,1 | ||
| Co | 0,1 | Mo | |||
| Cr | W |
Чаще всего лимитируется содержание железа и меди (допустимый уровень — 10–4–10–6%). Другие примеси определяют реже, но для некоторых веществ необходим контроль их содержания на уровне 10–7%, который не обеспечивается традиционными методами эмиссионной спектроскопии и спектрофотометрии. Особенно успешно каталитические методы применяются для определения Co, Mn, V, Mo, W, Nb, Ta. Кроме того, при анализе веществ особой чистоты каталитические методы позволяют определять отдельные анионы, органические соединения в неорганических солях, отклонения от стехиометрии в составе соединений.
Нижняя граница концентраций, определяемых с применением каталитических методов, для большинства элементов составляет 10–8–10–9 М (10–3–10–4 мкг/мл), т. е. на 1–2 порядка ниже, чем в методах спектрофотометрии, полярографии и пламенного варианта атомной абсорбции.
Скорость конкретной индикаторной реакции зависит от многих параметров: температуры, ионной силы раствора, концентрации мешающих компонентов, наличия ингибиторов и активаторов. Все эти параметры в кинетических методах анализа необходимо строго контрфолировать и поддерживать постоянными для получения надежной аналитической информации.
Каталитическая реакция является результатом протекания ряда элементарных процессов, поэтому влияние температуры на скорость процесса не всегда легко интерпретировать. Для практической оценки влияния температуры на скорость реакции используют температурный коэффициент , который показывает, во сколько раз увеличивается скорость реакции при возрастании температуры на 10 К:
Кинетическим определениям могут мешать посторонние ионы и соединения:
· обладающие каталитической активностью;
· снижающие скорость каталитической реакции или изменяющие направление ее отдельных стадий. Мешающее влияние оказывают окислители и восстановители, взаимодействующие с реагентами индикаторной реакции и катализатором: Mn(VII), Cr(VI), I2, , ,- ; анионы и соединения, образующие с катализатором каталитически неактивные комплексы — F-, HS , NH3, , ЭДТА; ионы переходных металлов, проявляющие каталитическую активность — Fe, Cu, Mn, Ti, Co; ионы металлов, образующие комплексы с реагентами и продуктами реакции — Al, Bi, Zr, Ti, TR и др.; ингибиторы и активаторы — в основном органические вещества, взаимодействующие с промежуточными продуктами реакции или с катализатором — оксалаты, фенолы, нафтолы, амины, а также инертные электролиты (например, нитраты и перхлораты щелочных металлов) и вещества, обладающие буферным действием.
Избирательность индикаторных реакций характеризуется максимально допустимым содержанием посторонних ионов, при которых относительная погрешность определения анализируемого вещества не превышает заданного значения, например, 0,1.
Кислотность раствора влияет на избирательность определения, если в лимитирующей стадии индикаторной реакции участвует ион водорода, а также в случае образования в результате гидролиза при разных значениях рН гидроксокомплексов, обладающих различной каталитической активностью. Так, каталитически активные комплексы Ti образуются при рН = 3,8, а максимальное содержание каталитически активных и наблюдается при рН = 1,1 и pH = 2,2, соответственно.
Для устранения мешающего влияния посторонних компонентов применяют традиционные для аналитической химии приемы отделения или маскирования мешающих примесей.
Кинетические методы (в том числе и ферментативные) при условии строгого соблюдения условий проведения анализа не уступают другим методам по точности, достаточно экспрессны, легко поддаются автоматизации. В практике аналитической химии эти методы применяют:
· при анализе смесей близких по свойствам органических соединений (некаталитический вариант);
· при определении микроколичеств ,- , Cl-ионов 3d-элементов, группы платиновых металлов, ряда анионов (I ) и органических веществ в природных и промышленных объектах;-Br
· при определении присутствия токсичных и лекарственных препаратов, физиологически активных соединений в биологических объектах и объектах окружающей среды (ферментативные методы).
· биохимические методы анализа, методы количественного определения неорганических и органических веществ, основанные на использовании процессов с участием биологических компонентов (ферментов, антител и др.). Аналитический сигнал - конечную концентрацию одного из продуктов реакции либо начальную скорость процесса, положенного в основу методики определения, регистрируют главным образом спектрофотометрическими, люминесцентными, электрохимическими методами. Наиболее распространены ферментативные и иммунохимические биохимические методы анализа.
· В ферментативных методах используют зависимость скорости химической реакции, катализируемой ферментом, от концентраций реагирующих веществ и фермента. Достоинства ферментативных биохимических методов анализа: высокая чувствительность, селективность, экспрессность и мягкие условия (25-37 °С, pH 5-10) проведения анализа. Предел обнаружения, нижняя и верхняя границы определяемых содержаний компонентов зависят от кинетических характеристик используемой индикаторной ферментативной реакции и, главным образом, каталитической активности фермента, от способа детектирования аналитического сигнала. Высокая селективность ферментативных биохимических методов анализа обусловлена образованием фермент-субстратного комплекса, требующим структурного соответствия субстрата активному центру фермента. С помощью ферментативных биохимических методов анализа определяют ферменты, субстраты, кофакторы и коферменты, активаторы и ингибиторы ферментов. Известны многочисленные высокочувствительные (нижние границы определяемых содержаний 10-9-10-6 моль/дм3) и селективные ферментативные биохимические методы анализа определения ионов металлов, неорганических анионов, пестицидов, фенолов, аминокислот, метаболитов, мутагенов, канцерогенов и других биологически активных соединений. Для создания биосенсоров, ферментных реакторов, разработки простых и экспрессных тест-методов определения токсикантов используют иммобилизованные ферменты. Высокая стабильность, возможность многократного использования иммобилизованных ферментов повышают экономичность, экспрессность анализа, позволяют его автоматизировать. Ферментативные биохимические методы анализа применяют в анализе объектов медицины (биологических жидкостей, тканей живых организмов), окружающей среды (природных и сточных вод, почв, воздуха, тканей растений), пищевых продуктов, фармацевтических препаратов, для непрерывного контроля микробиологических и биохимических процессов.
· Иммунохимические методы основаны на специфическом связывании определяемого соединения - антигена - соответствующими антителами. Малые концентрации комплекса антиген - антитело определяют, вводя в один из компонентов системы метку, детектируемую соответствующим инструментальным методом. Детектируемый сигнал пропорционален концентрации антигена. Используют изотопные, флуоресцентные, парамагнитные, ферментные метки. Анализ с использованием ферментной метки - иммуноферментный анализ - сочетает высокую чувствительность определения метки (предел обнаружения менее 10-12 моль/дм3) с уникальной специфичностью иммунохимического анализа. Иммунохимические биохимические методы анализа применяют для определения белков, пестицидов, гормонов, стероидов, наркотических и лекарственных средств, вирусов и клеток. Достоинства иммунохимического биохимического метода анализа: высокая чувствительность и специфичность определения, возможность использования малых объёмов анализируемой пробы (5-50 мкл). Время анализа - несколько минут. Разработаны диагностические иммуноферментные тест-системы для экспресс-определения биологически активных соединений в медицине, ветеринарии, микробиологической, фармакологической, пищевой промышленности, сельском хозяйстве, в объектах окружающей среды.
Электрохимические методы анализа основаны на измерении электрохимических свойств анализируемых систем, а именно на исследовании процессов, происходящих на электродах или в межэлектродном пространстве. При этом возникает или изменяется ряд параметров системы, таких как потенциал, ток, количество электричества, сопротивление, электропроводность и другие, значения которых пропорциональны концентрациям анализируемых веществ или определяются их специфическими свойствами. В основе классификации электрохимических методов положены процессы, происходящие на электродах.
1. Методы, основанные на протекании электродной электрохимической реакции: потенциометрия и потенциометрическое титрование, полярография, амперометрическое титрование, кулонометрия и кулонометрическое титрование.
2. Методы, не связанные с протеканием электродной электрохимической реакции. К ним относят кондуктометрию и кондуктометрическое титрование (низко- и высокочастотное).
Электрохимические методы анализа занимают важное место в технохимическом контроле пищевых продуктов. Широкое применение имеет потенциометрия и потенциометрическое титрование — для определения кислотности сырья, полуфабрикатов и готовой продукции, а также целого ряда ионов (Na+, Ca 2+, K+, NO 3-, I- и др.) на основе использования ионоселективных электродов.
Кондуктометрия используется для контроля водоочистки, для определения минеральных компонентов, в частности, например, в сахарном производстве для определения золы в диффузионных соках, уваренных сиропах, сахарном песке, патоке.
Очень широко используется полярография для определения токсичных металлов (Pb 2+, Cd2+, Zn2+, Cu2+и др).
Электрической проводимостью называют способность вещества проводить электрический ток под действием внешнего электрического поля. Электрическая проводимость электролитов обусловливается движением ионов под действием электрического поля.
Как и все проводники, растворы электролитов характеризуются определенным сопротивлением (R). Величина, обратная сопротивлению называется электрической проводимостью W
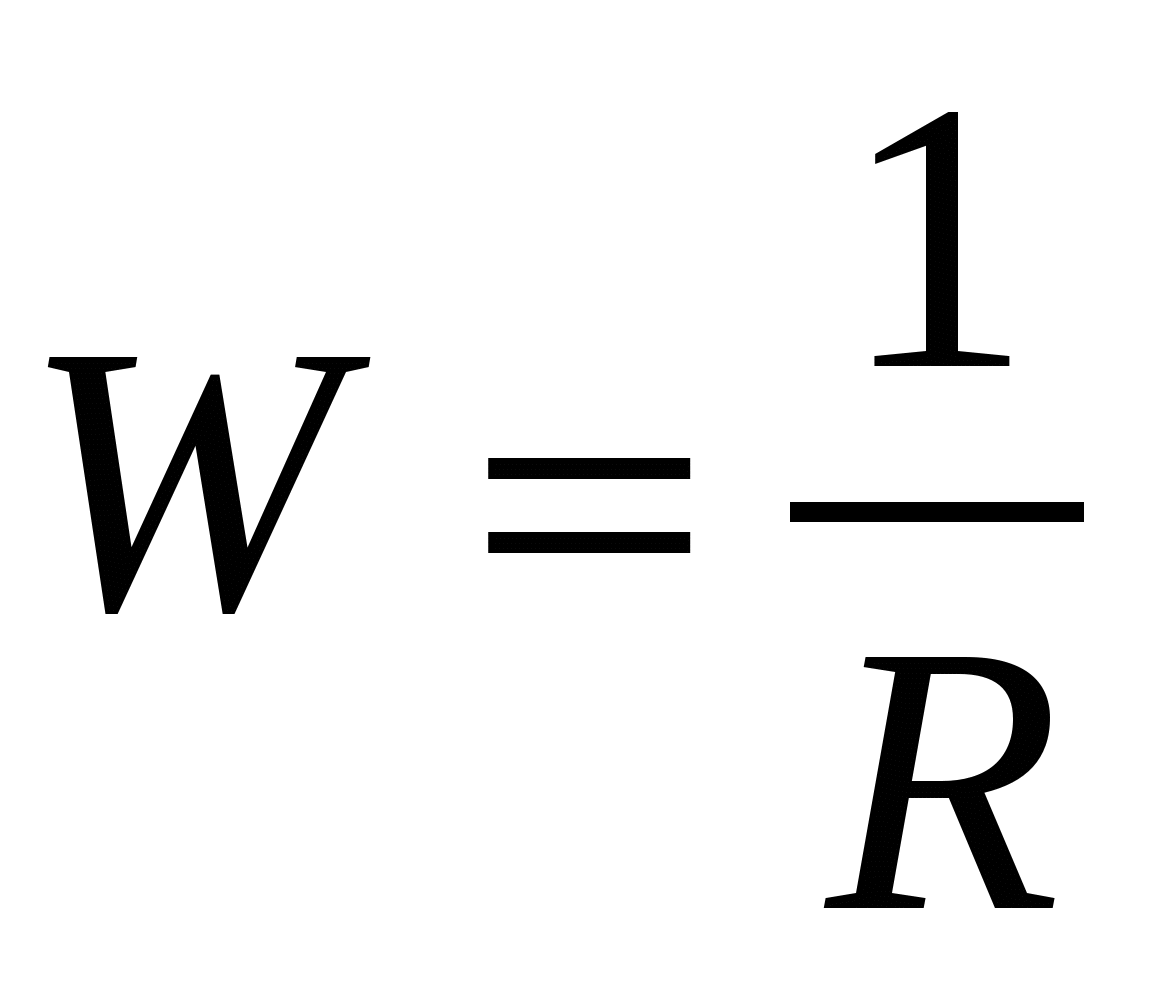 (Ом-1)
(Ом-1)
где: R — сопротивление раствора, Ом.
Единицей электрической проводимости является проводимость проводника сопротивлением 1 Ом. В системе СИ эта единица получила название сименс См.
Электрическая проводимость раствора выражается в единицах или удельной, или эквивалентной электрической проводимости. Удельная электрическая проводимость (χ) измеряется в См/м3и представляет собой электрическую проводимость 1 м3раствора, находящегося между параллельными электродами площадью 1 м2каждый при расстоянии между ними 1 м. Более удобной единицей объема для практического использования в лаборатории является дольная единица измерения, такая, как кубический сантиметр (см3). Тогда удельная электрическая проводимость будет измеряться в См/см3и представлять собой электрическую проводимость столба жидкости длиной 1 см м поперечным сечением 1 см2.
Для аналитических измерений более удобной характеристикой раствора электролита является эквивалентная электрическая проводимость λ. Эквивалентной электрической проводимостью называют проводимость раствора, содержащего 1 моль эквивалента вещества и находящегося между двумя параллельными электродами, расстояние между которыми 1 см. Ее единицей измерения, является См см2/моль-экв.
Удельная и эквивалентная проводимости взаимосвязаны соотношением
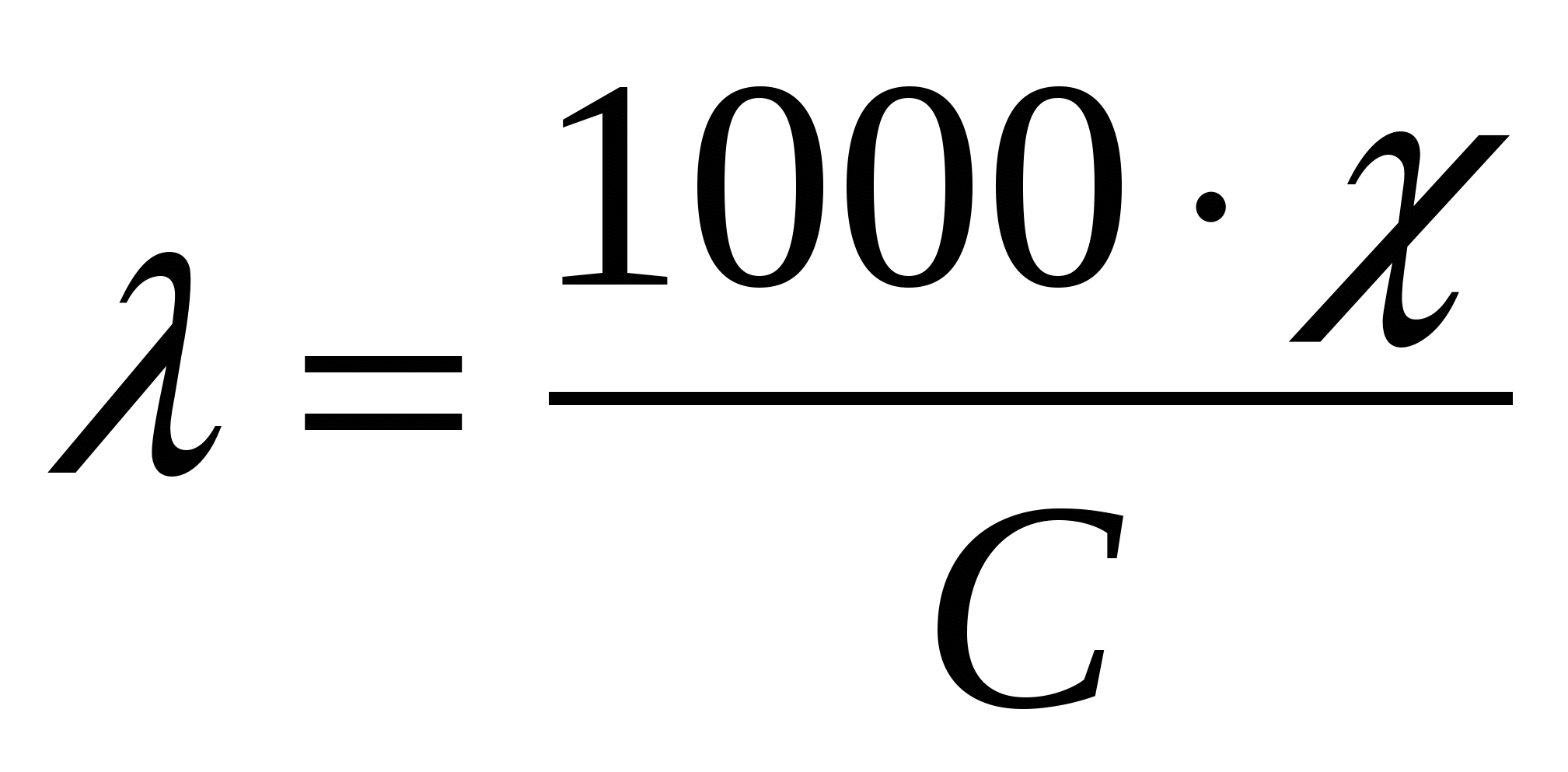
где: С- молярная концентрация эквивалента, моль/л.
Величина λ для сильных электролитов может быть выражена соотношением:
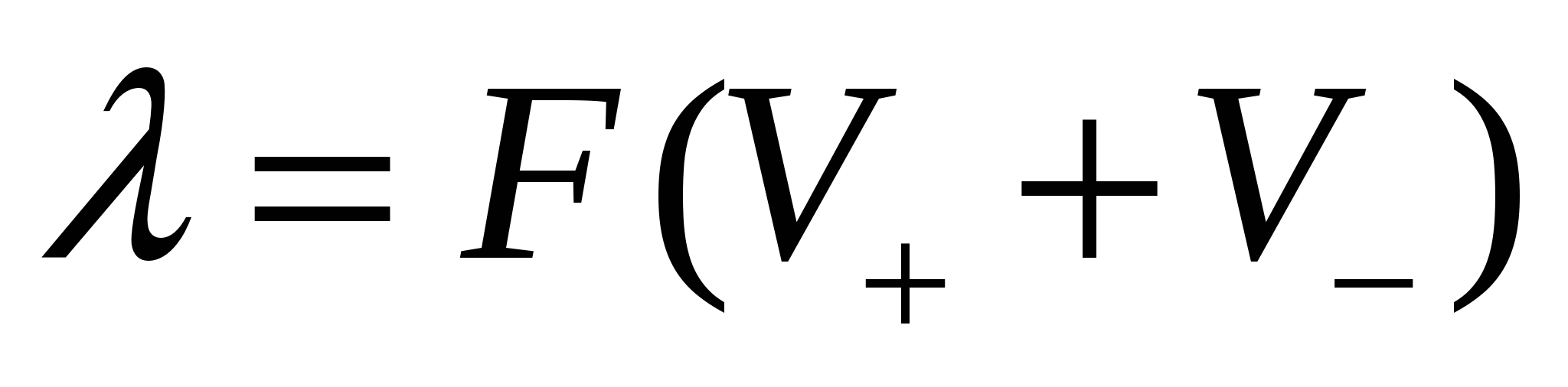
для слабых электролитов:
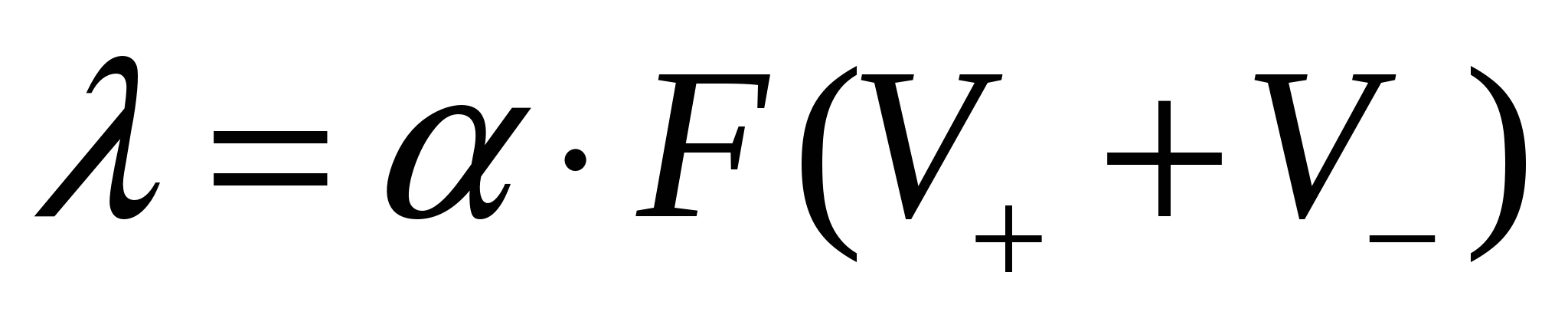
где: V+иV-— скорости движения катионов и анионов при градиенте потенциала 1 В/см, α — степень диссоциации электролита, F — число Фарадея (96500 Кл).
Произведение двух величин F∙V обозначают через U+ и U-, которые носят название подвижностей, или через λ+ и λ-, называемые эквивалентной электрической проводимостью ионов.
Числовые значения подвижностей ионов в водном растворе при комнатной температуре находятся в пределах 30-70 См см2/моль-экв и лишь у ионов H+ и ОH- они существенно превышают эти значения λ(Н+) = 350, λ (ОН-) = 199 СМ ∙см2/ моль-экв, что связано с особым механизмом перемещения этих ионов в электрическом поле.
1.2. Кондуктометрическое титрование
Различают прямую кондуктометрию и кондуктометрическое титрование. Метод прямой кондуктометрии основан на том, что в области разбавленных и умеренно концентрированных растворов электрическая проводимость растет с увеличением концентрации электролита. По величине электрической проводимости можно судить о концентрации раствора. В связи с относительно близкими значениями подвижностей ионов прямые кондуктометрические измерения дают информацию лишь об общей концентрации ионов в растворе. Малая селективность кондуктометрического метода является одним из его существенных ограничений. Метод прямой кондуктометрии применяют при контроле дистиллированной воды, для расчета констант диссоциации, для определения растворимости малорастворимых соединений.
Кондуктометрическое титрование основано на изменении в процессе титрования электрической проводимости исследуемого раствора, вызванном изменением концентрации ионов или заменой в растворе одного иона другим, обладающим иной подвижностью.
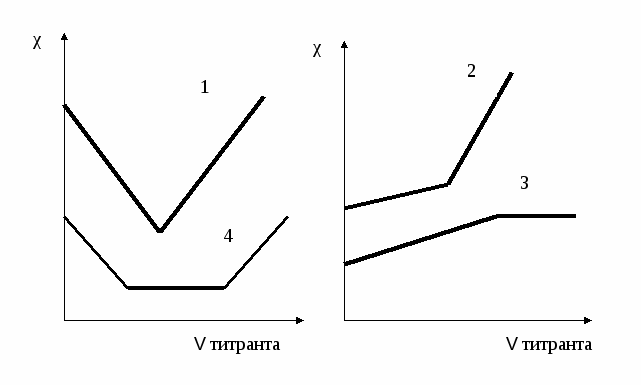
Результаты измерений электрической проводимости раствора в процессе титрования изображают графически в виде кривых титрования. Hа рис.1.1 приведены некоторые такие кривые титрования.
1). Титрование сильной кислоты сильным основанием. Hапример,
 H++ Cl-+ (K+ + OH-) → H2O + K++ Cl-.
H++ Cl-+ (K+ + OH-) → H2O + K++ Cl-.
При титровании в растворе происходит замена ионов водорода, имеющих большую подвижность, на менее подвижные ионы натрия или калия. При таком титровании электрическая проводимость раствора сначала уменьшается и достигает минимума в точке эквивалентности, после чего снова возрастает вследствие поступления в раствор избыточных ионов ОH-.
Рис.1.1. Кривые кондуктометрического титрования.
2). Титрование слабой кислоты сильным основанием. Hапример,
СH3СООH + Na++ OH- → CH3COO-+ Na++ Н2О
При титровании вместо слабодиссоциированной уксусной кислоты образуется ее натриевая соль, в процессе титрования электрическая проводимость несколько возрастает, а после окончания титрования при добавлении избытка сильного основания возрастает очень резко.
3). Титрование слабой кислоты слабым основанием. Hапример,
СHС3ООH + NH4OH → CH3COO- + NH4+ + Н2О
При титровании сначала электрическая проводимость увеличивается, так как образующаяся соль является сильным электролитом. После достижения точки эквивалентности электрическая проводимость практически не меняется.
4). Титрование смеси сильной и слабой кислот сильным основанием.
До точки нейтрализации сильной кислоты (первая точка эквивалентности) вследствие присутствия в растворе избытка сильной кислоты диссоциация слабой кислоты будет подавлена и вклад ее в общую электрическую проводимость раствора будет незначителен. Hа этом участке ход кривой титрования будет практически совпадать с ходом кривой титрования 1. Когда же вся сильная кислота будет нейтрализована, будет происходить титрование слабой кислоты, и кривая будет иметь вид кривой 2.
Область применения кондуктометрического титрования
Кондуктометрическое титрование дает хорошие результаты в тех случаях, когда электрическая проводимость исходного вещества значительно отличается от электрической проводимости продуктов реакции. Кондуктометрическое титрование может быть использовано для различных типов объемно-аналитических определений: кислотно-основных, осаждения, комплексообразования, обмена ионов и др.
Преимущества метода:
1. Возможность дифференцированного определения веществ в многокомпонентных смесях и водных растворах;
2. Возможность определений в окрашенных и мутных растворах, а также в присутствии окислителей или восстановителей, ограничивающих применение кислотно-основных индикаторов;
3. Возможность проводить определения не только в сравнительно концентрированных, но и разбавленных растворах (до 10-4М).
Ограничения. Метод имеет ограничения применения в тех случаях, когда в растворе присутствует много посторонних электролитов, т.к. электрическая проводимость раствора за их счет повышается и относительные изменения ее в ходе реакции титрования оказываются незначительными.
Высокочастотное титрование является видоизменением обычного кондуктометрического титрования.
В отличие от кондуктометрического титрования при высокочастотном титровании применяют не переменный ток небольшой частоты, а используют токи высокой частоты — от десятков тысяч герц до сотен мегагерц. Отличием высокочастотного кондуктометрического титрования от обычного кондуктометрического является также отсутствие непосредственного контакта исследуемого раствора с электродами.
Данный метод основан на регистрации изменения так называемой высокочастотной электрической проводимости G в процессе титрования. Величина G в свою очередь есть сложная функция от удельной электрической проводимости χ и частоты тока. Hа рис. 2.1. показана зависимость величины высокочастотной электрической проводимости G от удельной электрической проводимости при различных частотах тока для водного раствора КС1.
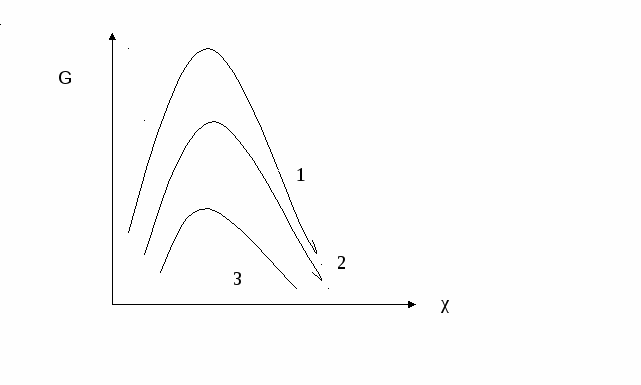
Рис. 2.1. Зависимость высокочастотной электрической проводимости G от удельной электрической проводимости
1 - 20 мГц, 2 - 10 мГц, 3 - 3 мГц.
Для осуществления метода высокочастотного титрования исследуемый раствор подвергается действию высокочастотного электромагнитного поля, создаваемого внутри измерительных ячеек, которые представляют собой либо электрический конденсатор, либо катушку индуктивности. По этому признаку измерительные ячейки делят на : 1) емкостные и 2) индуктивные.
Простейшая измерительная ячейка емкостного типа представляет тонкостенный стеклянный сосуд, на наружной поверхности которого монтируются две изолированные друг от друга металлические обкладки, подключенные к высокочастотному генератору.
Возможность помещать электроды снаружи сосуда - одно из главных преимуществ высокочастотного титрования. Переменный ток низкой частоты не может проходить через стенки стеклянного сосуда из-за большого емкостного сопротивления. С увеличением частоты сопротивление уменьшается. Если частота тока велика (что мы имеем при высокочастотном титровании), то через стенки сосуда и через раствор начинает протекать емкостной ток.
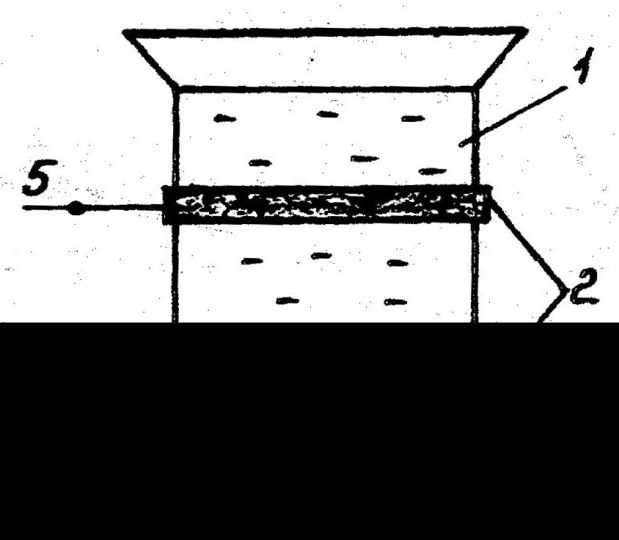
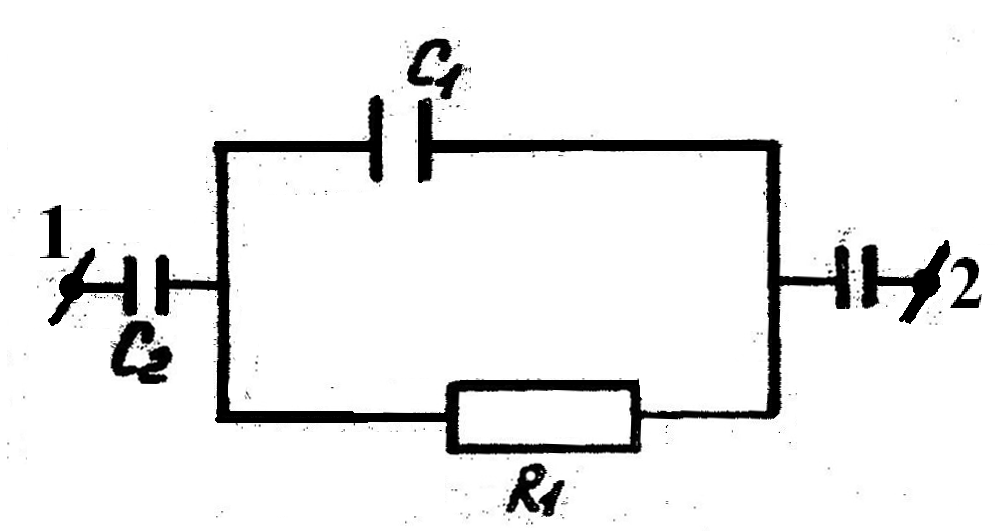
Измерительная ячейка емкостного типа представляет собой стеклянный цилиндрический сосуд, помещаемый между пластинами конденсатора (рис.2.2). Последние имеют форму колец. Учитывая, что раствор и стенки сосуда обладают собственными значениями диэлектрической проницаемости и электрическим сопротивлением, электрическую эквивалентную схему емкостной ячейки можно представить следующим образом (рис. 2.3).
| 
| ||
| Рис. 2.2 Высокочастотная измерительная ячейка емкостного типа: 1 - стеклянный сосуд, 2 - пластины конденсатора, 3 - исследуемый раствор, 4,5 - клеммы включения ячейки | Рис.2.3. Эквивалентная электрическая схема емкостной ячейки: R1 - сопротивление раствора, С1 - емкость раствора, С2 - емкость стенок сосуда и воздушного зазора между кольцами конденсатора и сосудом, 1,2 - клеммы ячейки | ||
При наложении высокочастотного напряжения к клеммам ячейки активное сопротивление R обусловливает определенную величину высокочастотной проводимости G, а следовательно, и определенную силу активного тока i .
а
б
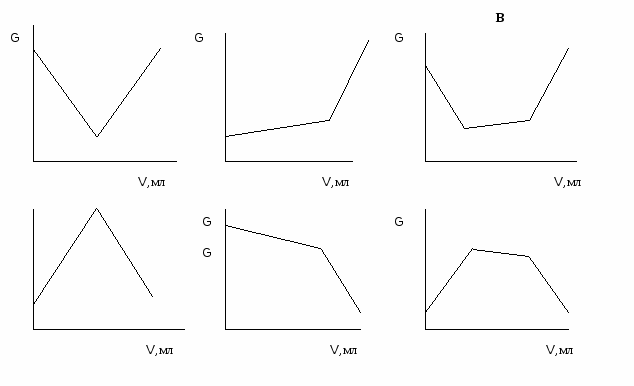
Р  ис. 2.4. Кривые высокочастотного кондуктометрического титрования
ис. 2.4. Кривые высокочастотного кондуктометрического титрования
Количество раствора, помещаемого в измерительную ячейку, должно быть таким, чтобы его мениск располагался выше верхней пластины конденсатора, иначе силовые линии электромагнитного поля могут замыкаться не только через жидкость, но и через воздух. Это приводит к резкому снижению чувствительности установки, кроме того, вследствие изменения уровня раствора при добавлении титранта показания индикаторного прибора титратора становятся функцией и высоты столба раствора. Поскольку при работе мешалки уровень жидкости колеблется, то при недостатке раствора возникают случайные нагрузки на измерительную ячейку, что влечет за собой хаотические броски стрелки индикаторного прибора.
В процессе титрования происходит изменение химического состава раствора, вызывающее изменение проводимости ячейки.
Hа рис. 2.4 изображены некоторые варианты кривых высокочастотного титрования соляной (а), уксусной (б) кислот и их смеси (в) основанием NaOH.
Вид экспериментально полученной кривой высокочастотного титрования зависит от пределов изменения концентраций исследуемого раствора(т.е. области изменения удельной электрической проводимости раствора в процессе титрования), значений высокочастотной проводимости (т.е. величины G) и частоты тока.
2.2. Практические работы
Работа 1. Определение хлороводородной и уксусной кислот
1. Аппаратура, материалы и реактивы
Титратор высокочастотный лабораторный ТВ-6Л-1 или другой марки
Стаканчик для титрования вместимостью 100 мл.
Пипетка вместимостью 5 мл.
Магнитный мешатель.
Бюретка вместимостью 25 мл.
Стандартный раствор HС1, 0,02500 М (готовят из фиксанала).
Раствор NaOH, 0,025 М.
2. Подготовка установки к работе
Прежде, чем приступить к выполнению работы необходимо:
1. Убедиться в том, что бюретка не подтекает, а носик ее заполнен раствором.
2. Опустить магнитный мешатель в стеклянный стаканчик и поместить его в датчик. Иметь в виду, что чувствительность прибора больше, если использовать для титрования стаканчик с тонкими стенками, плотно прилегающий к металлическим кольцам датчика.
3. Помнить, что во время титрования нельзя прикасаться к стаканчику или менять положение его в датчике.
4. Включить прибор в сеть на 220 В и включить тумблер "сеть". Прибор прогреть в течение 10 минут.
3. Выполнение работы
Титрование растворов кислот основано на реакции нейтрализации
HС1 + NaOH = NaCl + H2О
или
CH3COOH + NaOH = CH3COONa + H2О
Титрант — NaOH.
Дата добавления: 2016-02-27; просмотров: 4427;