ПЕРЕДАЧА ЭЛЕКТРОНОВ
Львиная доля производимой в процессе обмена веществ энергии выделяется в ходе реакций, в которых участвует атмосферный кислород. Перед тем как мы сможем свободно рассуждать об этих реакциях, надо сначала обговорить основные вещи.
Общепринятый термин для описания всех реакций, при которых в наличии имеется пламя, – «горение». Однако, после того, как Лавуазье показал, что при сгорании древесины происходит массированное соединение содержащихся в ней веществ с кислородом, распространение получило и более осмысленный термин «окисление».
Этот термин оказался не только более осмысленным, но и более общим. Процесс медленного соединения металлов с кислородом, в результате которого образуется окалина (сейчас мы называем ее оксидом), как показал Лавуазье, тоже является окислением, хотя горением его не назовешь. Иными словами, окисление – это любая реакция соединения вещества с атмосферным кислородом, независимо от того, вспыхивает ли при этой реакции пламя. Атмосферный кислород является в данном случае примером «агента окисления» (это может показаться тавтологией, но вскоре я покажу, что атмосферный кислород – не единственный распространенный в природе агент окисления).
С другой стороны, долгое время было принято говорить о процессе «восстановления» (англ. «reduction», от латинского «вести обратно») руды в состояние металла. Люди много веков наблюдали, как железо превращается в ржавчину, а большая часть металлических руд тоже похожа на ржавчину.
Обращаться с оксидами металлов, как с рудой, приходится довольно часто. Чтобы снова сделать из них металл, требуется удалить из состава оксидов атом кислорода. Так, если нагревать железную руду вместе с коксом, то атомы углерода, содержащиеся в коксе, соединятся с атомами кислорода, содержащимися в руде, оставляя металл свободным:
2Fe2O3 + ЗС → 3CO2 + 4Fe.
Поскольку восстановление спровоцировал углерод, то он являет собой пример «агента восстановления».
Таким образом, окисление – это процесс добавления кислорода к веществу, а восстановление – процесс удаления кислорода из вещества. Природа этих двух процессов прямо противоположна.
Есть еще один распространенный способ устранения кислорода из соединения с металлом – обработка водородом. В этом случае водород, как и углерод, служит агентом восстановления. Он вступает в соединение с кислородом и восстанавливает, в частности, медь, после того как она окислится дочерна.
Реакции с использованием водорода вызывают особый интерес у органических химиков. Они обнаружили, что определенные химические вещества, богатые кислородом и способные окислять металлы путем формирования оксидов, имеют также и свойство «выдирать» водород из молекул органических соединений. Соответственно, эти богатые кислородом вещества, среди которых – перманганат калия (КMnO4) и хромовокислый калий (К2CrO4), тоже являются агентами окисления. У ученых создалось стойкое представление, что все они отдают часть своего кислорода на окисление металлов или используют его для соединения с водородом, выцепляя его из органических молекул с последующим образованием воды. Соответственно, потеря водорода (дегидрогенизация) тоже стала рассматриваться учеными как форма окисления, даже если ни один атом кислорода к органической молекуле напрямую и не присоединялся.
Однако, если окисленную таким образом органическую молекулу обработать газообразным водородом (как правило, в присутствии какого‑нибудь катализатора), реакцию окисления можно обратить вспять. Добавление водорода (гидрогенизацию) можно, таким образом, рассматривать как реакцию восстановительную, даже если опять же никакой кислород ниоткуда не извлекался.
Короче говоря, органические химики решили рассматривать окисление как добавление кислорода или удаление водорода, а восстановление – как удаление кислорода или добавление водорода.
Но и это еще не все обобщение. Металлы покрываются ржавчиной и патиной и под воздействием фтора, хлора, брома и серы, хотя ни кислород, ни водород тут вообще ни при чем. Точно таким же образом в качестве восстановительных агентов могут выступать металлический натрий, калий, магний и кальций, хотя в происходящих с их участием реакциях не затрагивается ни кислород, ни водород. Распространять термины «окисление» и «восстановление» на бесконечное множество реакций добавления и извлечения того или иного вещества казалось уже неразумным, и надо было ввести какое‑то более глобальное обобщение. Оно было сделано только после открытия электронов.
В XX веке обнаружилось, что, подвергаясь окислению, вещество теряет электроны, а подвергаясь восстановлению – получает. Возникло естественное предложение вообще перестать использовать термины «окисление» и «восстановление» в связи с приобретением или потерей веществом какого‑либо элемента, а пользоваться ими для обозначения процессов потери или приобретения электронов.
К примеру, когда металлический натрий вступает в реакцию с газообразным хлором для образования хлорида натрия, атом натрия теряет электрон и становится ионом натрия. Соответственно, можно сказать, что хлор окислил натрий, и рассматривать хлор как агент окисления. Однако в ходе той же самой реакции атом хлора приобретает электрон и становится ионом хлора. Значит, натрий восстанавливает хлор и является в таком случае агентом восстановления (рис. 53).
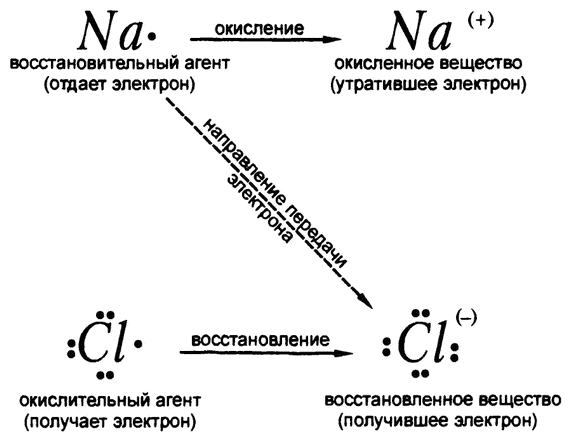
Рис. 53. Окислительно‑восстановительная реакция
Опять же, при горении углерода в кислороде образуется углекислый газ, в котором каждый атом углерода связан ковалентными связями с двумя атомами кислорода. Передачи электрона, как таковой, в данном случае не происходит, ни один атом не теряет полностью электрона и не приобретает его. Но атом кислорода держит электроны крепче, чем атом углерода. Так что можно сказать, что атом углерода теряет часть своего электрона и все же является окисленным, а кислород выступает в качестве агента окисления. С другой стороны, атом кислорода приобретает часть электрона, а значит – восстанавливается, а восстановительным агентом является углерод.
Как видите, не бывает ни окисления самого по себе, ни восстановления самого по себе. В обычных химических реакциях электроны не могут существовать сами по себе, они могут лишь переходить от одного атома к другому, целиком либо частично. Атом, теряющий электрон, – окисляется, приобретающий – восстанавливается. Значит, окисляемое вещество всегда является агентом восстановления для второго участвующего в реакции вещества, а то, в свою очередь, – агентом восстановления для первого. Разумеется, это касается не всех реакций – если атомы распределяются поровну, как в случае образования молекулярного хлора из атомного, то в этом процессе нет ни восстановления, ни окисления.
Принимая во внимание неразрывную связь между окислением и восстановлением, химики дали такого рода реакциям двойное название «окислительно‑восстановительных».
Так как же проходят с точки зрения перераспределения электронов реакции гидрогенизации и дегидрогенизации?
Эффект от потери водорода необходимо рассматривать по‑разному, в зависимости от того, имела ли место также потеря электронов, поскольку для данного процесса это вопрос первостепенной важности. Иногда вещество высвобождает атом водорода вообще без «положенного» ему электрона. Водород, лишенный электрона, является положительно заряженным ионом водорода Н+. А остаток вещества становится, таким образом, отрицательно заряженным ионом, и весь процесс в целом получает название ионизации. Получило ли вещество в этом случае электрон и можно ли считать его подвергшимся «восстановлению»?
Правильный ответ – нет. Ион водорода высвобождается только в том случае, если его связь с неким совместно используемым электроном (общим, чаще всего, с кислородом) уже является столь слабой, что он просто «отпадает» от соединения. Он не теряет электрона в процессе ионизации, а остальная часть соединения – не приобретает его. Скорее следует признать факт свершившейся ионизации признаком того, что электрон уже перешел от водорода к остальной части вещества в результате каких‑то предыдущих событий. Сама по себе ионизация не является ни окислением, ни восстановлением.
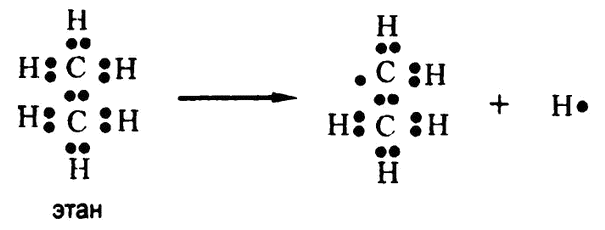
Рис. 54. Дегидрогенизация – электронная запись
Но, предположим, что атом водорода удален из соединения вместе со своим электроном. Такая ситуация представлена на рис. 54. (Вопрос о том, что происходит при этом с самим атомом водорода, отложим на потом. Сейчас рассмотрим только освободившуюся связь.)
Теперь можно сказать, что у органического вещества остался один электрон в полном распоряжении – а раньше было два, но в частичном. Поскольку атом углерода держит электроны чуть крепче, чем атом водорода, то когда у углерода и водорода было два совместно используемых электрона, на долю углерода приходилась чуть большая часть. Теперь же «чуть большая часть от двух», то есть «чуть более одного электрона», заменено на просто «один электрон». Значит, часть электрона соединение потеряло, и его можно счесть окисленным. Таким образом, дегидрогенизация остается окислительной реакцией и в новом, электронном понимании.
Следуя такой же логике, можно доказать и что потеря атома кислорода (хоть в одиночку, хоть в составе гидроксильной группы) вместе со всеми электронами предоставляет оставшейся части молекулы полный контроль над одним электроном вместо слабых попыток совместного с кислородом контроля над двумя электронами. Оставшаяся часть молекулы приобретает таким образом долю электрона, так что удаление из нее кислорода означает восстановление.
Но вернемся к дегидрогенизации, схема которой приведена на рис. 54. Обратите внимание, что после удаления атома водорода у углевода остается один непарный электрон. Группа атомов, обладающая непарным электроном, называется «свободным радикалом».
Свободные радикалы в таком виде пребывают недолго, поскольку оставшийся без пары электрон – очень нестабильное образование. Он вынужден искать себе пару, и вскоре обязательно с его участием происходит какая‑нибудь химическая реакция.
Конечно, если удалить два атома водорода (каждый вместе со своим электроном), так что одновременно в одной молекуле образуются два непарных электрона, то они вполне могут найти себе пару в лице друг друга, образовав «двойную связь», как на рис. 55.
В большинстве случаев окисления органических веществ происходит действительно именно такая «двойная» дегидрогенизация – потеря двух атомов водорода. При такого рода дегидрогенизации происходит значительное снижение уровня свободной энергии – настолько значительное, что такие реакции, происходящие в процессе катаболизма, и представляют собой источник энергии для образования высокоэнергетических фосфатных связей.
Тем не менее при обычных условиях такого, как правило, не случается. Органическое вещество может существовать годами, и никакой дегидрогенизации с ним не происходит, пока оно не окажется в центре значительного изменения уровня свободной энергии.
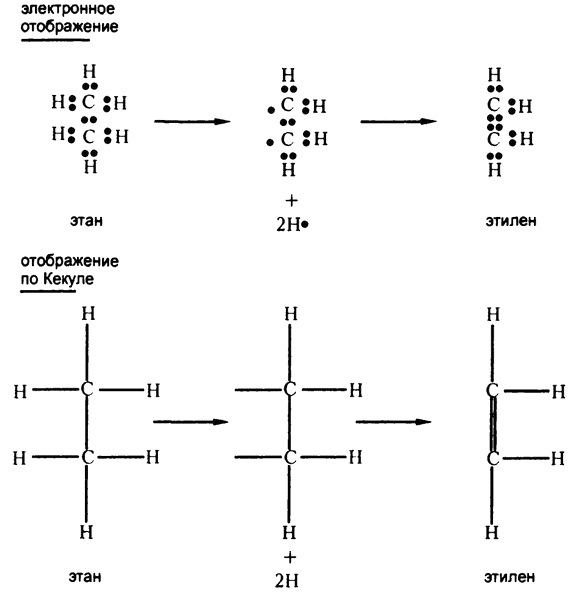
Рис. 55. Образование двойных связей
Понятно, что потребуется еще и значительное количество энергии активации. В 1935 году Михаэлис (тот самый, один из авторов формулы Михаэлиса– Ментена) указал, что крайне маловероятно, чтобы органическое соединение потеряло два атома водорода одновременно. Гораздо вероятнее, что сначала оно потеряет один атом, а потом – второй. То есть должна существовать некая промежуточная форма, когда один электрон уже утрачен, а второй – еще нет, и в этот момент вещество будет представлять собой свободный радикал.
Свободный радикал – нестабилен, а для его создания требуется приложить много свободной энергии. С потерей второго электрона энергозатраты более чем восполнятся; однако сначала все же надо оторвать первый электрон, а для этого надо найти энергию активации, и именно эта сложность и не дает всем органическим соединениям (включая те, из которых состоим мы сами) немедленно разложиться на воду и углекислоту.
Однако в тканях имеются ферменты, катализирующие дегидрогенизацию при комнатной температуре. Это делается за счет снижения энергетического содержания свободного радикала – его стабилизации, иными словами. Таким образом, энергия активации уменьшается до той точки, при которой кинетической энергии молекул при комнатной температуре оказывается достаточно, чтобы приводить к образованию свободных радикалов. Когда это происходит, то, разумеется, тут же теряется и второй атом водорода и образуется двойная связь.
Но каким же образом стабилизируется свободный радикал?
На протяжении всего XIX века химики пытались создать вещества, которые мы сейчас называем свободными радикалами и которые существовали бы хоть сколько‑нибудь долго, но ничего у них не выходило. Получилось это, наконец, у американского биохимика Мозеса Гомберга только в 1900 году. Гомберг показал, что некоторые сложные свободные радикалы можно заставить существовать неопределенно долго, и об их существовании свидетельствует цвет раствора.
Первый созданный таким образом свободный радикал, трифенилметил, состоит из атома углерода, соединенного с тремя бензольными кольцами. (Сам бензол состоит из шести атомов углерода и шести атомов водорода, объединенных в симметричное замкнутое кольцо.) Все три бензольных кольца, как нам известно, находятся в одной плоскости и расположены симметрично относительно центрального атома углерода, как изображено на рис. 56.
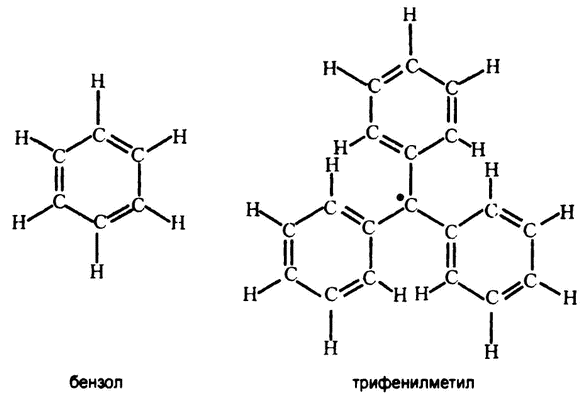
Рис. 56. Свободный радикал трифенилметил и его отношение к бензольному кольцу
Но с чего вдруг этот конкретный свободный радикал оказался таким стабильным? Впервые разумно объяснить это смог американских химик Лайнус Полинг только в начале 30‑х годов XX века. Полинг взял на вооружение теории 20‑х годов о том, что электрон – это не крошечный шарик, лишенный собственных свойств, а совокупность волн, способных размываться по большему или меньшему пространству, в зависимости от обстоятельств.
Основываясь на представлении о волновой природе электронов, Полинг разработал математическую теорию, которую назвал «теорией резонансов». Помимо прочего, с помощью этой теории он смог показать, что чем в большей степени непарный электрон способен размываться, тем более он стабилен.
Вероятность размывания электрона больше в тех случаях, когда молекула, в состав которой он включен, расположена в одной плоскости и симметрична. Трифенилметил Гомберга идеально удовлетворяет этим требованиям. На рис. 56 я изобразил непарный электрон в виде точки, но это уже не соответствует реальным представлениям химиков о природе электрона. Правильнее было бы изобразить его в виде‑туманности, обволакивающей молекулу, с симметричными областями высокой концентрации на некоторых участках.
Михаэлис, услышав про теорию резонанса, указал, что соединение фермента с субстратом – гораздо более симметрично, чем молекула субстрата сама по себе. В таком случае свободный радикал субстрата, объединенного с ферментом, будет гораздо более устойчивым соединением, чем просто свободный радикал субстрата, и для образования свободного радикала из комплекса «субстрат‑фермент» потребуется гораздо меньше энергии, чем для образования свободного радикала из просто субстрата, – именно за счет этого фермент столь успешно катализирует реакцию.
Вернемся же к судьбе тех двух атомов водорода, которые были удалены из органического вещества путем дегидрогенизации. Один из возможных вариантов – что они объединятся в молекулу газообразного водорода и улетят. Однако при дегидрогенизации органических веществ в живой ткани этого, как правило, не происходит, за исключением тканей нескольких видов бактерий.
Альтернативный вариант – атомы перейдут к некоей «принимающей» молекуле, которая, приобретя атомы водорода, таким образом «восстанавливается». В качестве варианта такой молекулы напрашивается кислород, значит, пора нам присмотреться к электронному строению этого элемента.
В атоме кислорода восемь электронов, два во внутренней оболочке и шесть – во внешней. В электронной записи этот атом можно отобразить так:

В обычных условиях этот атом в свободном виде не встречается, а существует по парам, составляющим молекулу кислорода. Логично предположить, что при этом у двух атомов формируется общее множество из двух пар электронов, так что в оболочке каждого из них оказывается вожделенная восьмерка:

Проблема в том, что кислород обладает, ко всему прочему, еще и сильным магнетическим действием, и химики уверены, что причиной тому – наличие непарных электронов. Так что более вероятной кажется вот такая картина:

Это уже два свободных радикала, которые тем не менее являются стабильными. Достаточно интересное исключение для тех, кто занимается теоретической химией, но мы сейчас в это углубляться не будем.
Такая молекула кислорода легко может принять два атома водорода следующим образом (привожу как «электронную», так и обычную запись):

H2O2 – это перекись водорода, достаточно нестабильное соединение, имеющее тенденцию к расщеплению на воду и кислород:
2H2O2→ 2H2O + O2.
Распад перекиси водорода – спонтанная реакция, представляющая уменьшение свободной энергии (рис. 57). Своей медленной скоростью (а хранить раствор перекиси водорода при низких температурах можно достаточно долго без значительных потерь) она обязана тому факту, что для распада необходимо разорвать связь О–О, а для этого нужна достаточно большая энергия активации.
Для получения этой энергии активации, а значит, и ускорения распада перекиси водорода достаточно даже слабого нагревания. Ряд химических веществ способны этот распад катализировать, например железные опилки или широко распространенный фермент каталаза.
Поскольку при переходе от перекиси водорода к воде уровень свободной энергии значительно снижается, то можно сказать, что при переходе от кислорода к воде уровень свободной энергии снижается сильнее, чем при переходе от кислорода к перекиси водорода. Следует ли в таком случае ожидать, что кислород, принимая водород, каждый раз будет образовывать воду?
Застревание происходит на этапе разрыва связи О–О. Если кислород соединяется с водородом при высоких температурах (к примеру, при сжигании смеси кислорода с водородом или сжигании же какого‑нибудь органического соединения) связь кислорода с кислородом рвется без проблем и образуется вода.
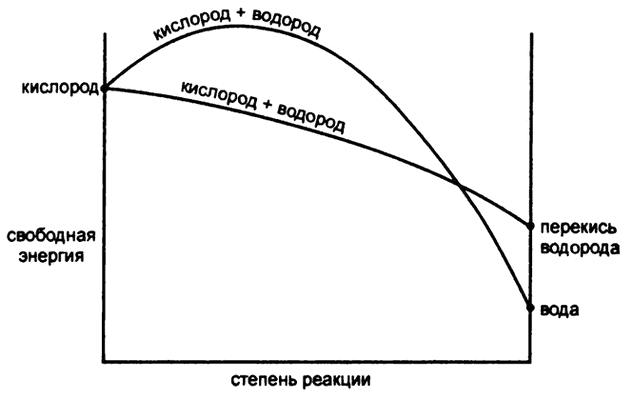
Рис. 57. Перекись водорода и вода
Если же кислород соединяется с водородом при комнатной температуре, как это случается при некоторых реакциях, катализируемых ферментами, на разрыв связи «кислород‑кислород» энергии может и не хватить и вместо воды образуется перекись водорода.
Однако в живых тканях перекись водорода в каких‑либо значительных концентрациях не встречается – и хорошо, потому что она очень ядовита. Одним из возможных тому объяснений служит присутствие каталазы. Ее действие может заключаться в том, что любая образующаяся молекула перекиси водорода тут же расщепляется с образованием воды. Однако, как мы вскоре увидим, вполне вероятно, что крупномасштабного образования перекиси водорода в организме вообще не происходит, а каталаза служит лишь для подстраховки и потребность в ней возникает нечасто.
Кислород, конечно, наиболее вероятный захватчик свободного атома водорода, но не единственно возможный. Атом водорода, который одно органическое вещество отпускает, может быть принят целым рядом других органических веществ.
Чтобы лучше понять этот факт, представим, что множество органических веществ могут существовать в двух формах – в восстановленной и окисленной, причем разница между ними будет заключаться в присутствии или отсутствии двух атомов водорода. Это можно выразить следующим образом:
АН2 ↔ А + 2Н.
ВН2 ↔ В + 2Н и так далее.
восстановленная форма ↔ окисленная форма
Окислительно‑восстановительные реакции будут, естественно, разными для разных веществ. К примеру, у первой из вышеприведенных реакций точка равновесия может быть смещена вправо, так что АН2 будет иметь ярко выраженную склонность к потере двух атомов водорода. С другой стороны, у второй реакции точка равновесия может быть смещена влево, так что В будет иметь склонность к присоединению атомов водорода.
В таком случае, если смешать АН2 и В, то получим следующую реакцию:
АН2+ В → ВН2 + А.
АН2, теряя атомы водорода, окисляется агентом окисления В. Само же В, приобретая водород, восстанавливается агентом восстановления АН2.
Теперь нам остается только разработать какой‑то способ измерить склонность водорода перетекать от одного вещества к другому, и это даст нам возможность предсказать, какое из них будет окисляться, а какое – восстанавливаться.
Для этого необходимо вспомнить, что и потеря, и принятие атомов водорода на самом деле лишь один из аспектов потери и принятия электронов. Любое вещество, имеющее склонность раздавать электроны, оказывает «давление», посылающее их по электрической цепи. Такое давление является электрическим потенциалом. Можно установить электрические батареи типа I, о которых я говорил в главе 10, и измерить в вольтах электрический потенциал той или иной окислительной реакции. Привести их к определенным стандартным условиям – и мы получаем «окислительный потенциал».
Окислительный потенциал реакции, при которой атом водорода отдает один электрон и превращается в ион водорода, произвольно принят за ноль. Окислительный потенциал любого атома или молекулы, проявляющих меньшую склонность к тому, чтобы отдавать электрон, считается положительным, проявляющих большую склонность отдать электрон – отрицательным.
Любое вещество всегда будет принимать электроны (иногда вместе с атомами водорода в качестве бесплатного приложения) от другого вещества, имеющего меньший положительный (или вообще отрицательный) окислительный потенциал. И точно так же любое вещество будет отдавать электроны (иногда вместе с атомами водорода) другому веществу, имеющему больший положительный (или меньший отрицательный) окислительный потенциал.
Можно даже представить серию окислительно‑восстановительных реакций с участием одних и тех же электронов или одних и тех же атомов водорода, переходящих от одного вещества к другому по цепочке последовательного уменьшения отрицательных и увеличения положительных окислительных потенциалов.
Именно так на самом деле и происходит в живой ткани – давайте же вернемся к ней.
Глава 23.
ЖИЗНЬ С ВОЗДУХОМ
При рассмотрении реакций, проходящих с участием атмосферного кислорода, естественно возникает желание разобраться в самом процессе впитывания кислорода живой тканью (ну, наполняет он легкие, и что дальше?).
Из таких разных существ, как картошка и мышь, добывались образцы тканей, системы ферментов в которых оставались в достаточной степени нетронутыми, чтобы продолжать катализировать положенные им реакции с использованием кислорода даже в отрыве от самого организма. Для некоторых целей нет необходимости сохранять клетки нетронутыми, поскольку реакции, потребляющие кислород, могут происходить и в достаточно чистых растворах ферментов.
Такие реакции обычно проводят в маленьком закрытом контейнере, так чтобы изменения давления в нем оказывались ощутимыми. Такой контейнер состоит из двух частей, в одной из которых находится раствор субстрата, а в другой – раствор фермента или образцы тканей. Стоит чуть‑чуть покачать контейнер – и начнется реакция.
В обычных условиях начал бы потребляться кислород и выделяться углекислый газ. В результате потребления кислорода в контейнере мог бы образоваться частичный вакуум, но благодаря производству углекислого газа (что всегда происходит, когда речь идет о живых клетках) потеря возмещается и нормальное давление газа сохраняется. Однако, если внутри контейнера есть еще и небольшая ниша, в которой находится некий раствор, быстро поглощающий углекислый газ, то возмещения общего объема газа в контейнере происходить не будет. Общее снижение давления будет свидетельствовать о потреблении кислорода.
Контейнер герметично соединен с U‑образной трубкой, открытой в атмосферу, нижняя часть которой заполнена цветной жидкостью. С одной стороны трубки на жидкость давит воздух, содержащийся в контейнере, с другой – обычное атмосферное давление. По мере протекания в контейнере катализируемой ферментом реакции и потребления кислорода давление в нем падает, и баланс между внутренним и внешним давлением нарушается. Жидкость в трубке со стороны атмосферы ползет вниз, а со стороны, присоединенной к контейнеру, – вверх.
Естественно, в таких случаях принимаются все меры к тому, чтобы движение жидкости осуществлялось только под воздействием возрастающей разреженности воздуха в контейнере. Контейнер помещен в теплую ванночку и содержится при постоянной температуре во избежание изменения объема воздуха в связи с изменениями температуры. Рядом находится такой же контейнер, но лишенный фермента, – контрольный, чтобы экспериментаторы видели, насколько на колебания жидкости в трубке влияют посторонние факторы, например небольшие колебания атмосферного давления.
В конечном итоге точно фиксируется сдвиг уровня жидкости в трубке и по нему определяется количество потребленного в результате катализируемой ферментом реакции кислорода. Только что описанное устройство в самом распространенном виде изобрел в 1923 году немецкий биохимик Отто Варбург, поэтому его иногда называют «респирометром Варбурга». Слово «респирометр» происходит от слов «дыхание» и «мерить».
Если в респирометр Варбурга поместить растолченную мышцу, то по мере того, как содержащиеся в ней ферменты будут катализировать реакции, кислород будет потребляться. Однако через некоторое время потребление кислорода начинает снижаться до весьма низких значений. Казалось, что ферменты денатурировались и утратили активность. Впрочем, потеря активности наблюдалась и в том случае, когда ферментам создавались все условия для того, чтобы они не денатурировались. Тогда возникло предположение, что замедление реакции связано с полной выработкой субстрата.
В последнем случае потребление кислорода снова возросло бы с добавлением субстрата, – оставалось только решить, что же именно является необходимым субстратом. Видимо, субстрат для некоторых ферментов находился не в растворе, помещенном в контейнер, а в самой мышце, так что в контейнер стали по очереди добавлять обнаруживаемые в ней вещества.
В 1935 году венгерский биохимик Альберт Сент‑Дьёрдьи установил, что потребление кислорода возобновляется в том случае, если к толченой мышце добавить одно любое из четырех веществ – янтарную кислоту, фумаровую кислоту, яблочную кислоту или щавелево‑уксусную кислоту. Молекулярное строение этих веществ сходно, как видно из рис. 58. Все они являются «двухосновными кислотами», поскольку молекула каждой содержит по две карбоксильных группы.
Очевидно, каждое из этих веществ каким‑то образом задействуется в реакциях с потреблением кислорода. Однако они не могут использоваться просто как строительный материал, поскольку объем кислорода, который начинает потребляться после добавления любого из этих веществ, значительно превышает необходимый для связывания всего вновь добавленного количества. Видимо, начинает происходить не одна реакция, а ряд реакций, при которых используемое вещество (к примеру, янтарная кислота) возобновляется с той же скоростью, что и расходуется, так что ее участие скорее следует счесть каталитическим. По мере продолжения серии реакций становится понятно, что запас янтарной кислоты возобновляется не полностью, так что идет ее медленное растрачивание, и, в конце концов, для продолжения реакций оказывается необходимым добавление новой порции янтарной кислоты извне.
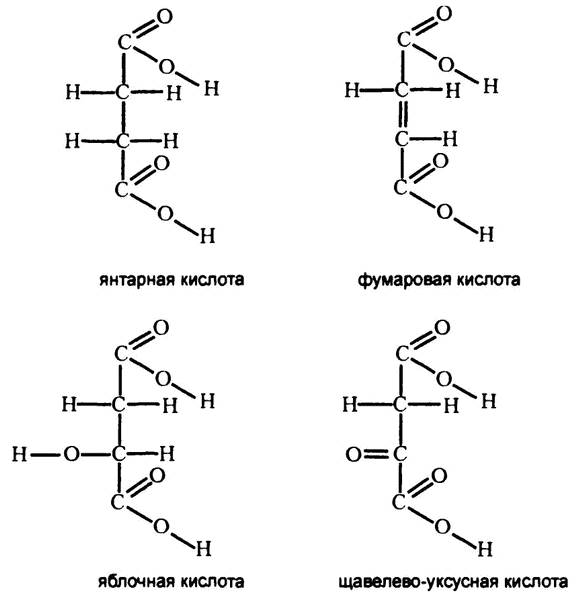
Рис. 58. Двухосновные кислоты
Дальше мы увидим, что четыре перечисленных вещества принимают участие не в четырех различных реакциях, а в одной и той же. Доказательство этого факта было получено благодаря малоновой кислоте.
Если в толченую ткань добавить янтарной кислоты для того, чтобы возобновить потребление кислорода, то стоит только после этого добавить малоновой кислоты, как потребление кислорода тут же снова прекращается. Это вполне ожидаемый эффект, поскольку малоновая кислота является конкурентным угнетателем янтарной (см. главу 18). Однако после добавления малоновой кислоты останавливалось потребление кислорода и возобновившееся после добавления любого из трех остальных веществ. А ведь напрямую подавлять реакции, проходящие с участием яблочной или щавелево‑уксусной кислоты, малоновая кислота не может. Значит, будучи добавленной в растолченную ткань, она останавливает общий ход реакции за счет того, что где‑то в нем используется часть именно янтарной кислоты. После того как было проведено много исследований в этом направлении, немецкий биохимик Ханс Адольф Кребс сумел продемонстрировать, что добавление и других веществ, в частности – лимонной кислоты (в которой содержится шесть атомов углерода и три карбоксильные группы, а не четыре атома углерода и две карбоксильные группы, как в четырех установленных Сент‑Дьёрдьи кислотах), тоже приводит к возобновлению реакций с потреблением кислорода. В 1940 году Кребс разработал схему, в которой все необходимые вещества заняли свое логическое положение, и с тех пор эта схема претерпевала лишь самые незначительные изменения.
По понятным причинам эта серия химических реакций получила название «цикл Кребса», но есть названия и более описательные – «цикл трикарбоновых кислот», или «лимоннокислый цикл», поскольку лимонная кислота обладает тремя карбоксильными группами.
Давайте посмотрим, стараясь обойтись как можно меньшим числом формул и подробностей, как же работает цикл Кребса.
При анаэробном гликолизе, как я объяснял в главе 20, молекула глюкозы превращается в две молекулы молочной кислоты, производя при этом две высокоэнергетические фосфатные связи, принимающие в итоге вид АТФ. Далее предстоит катаболизм молочной кислоты с образованием дополнительных высокоэнергетических связей.
Этот процесс начинается с устранения из молочной кислоты двух атомов водорода (дегидрогенизации) с образованием пировиноградной кислоты, как показано на рис. 59.
Сочетание С=О называется «кетоновой группой», или, короче, «кетогруппой», а сочетание СООН, вам, я надеюсь, уже знакомое, – «карбоновокислой группой». Любое химическое соединение, где, как, например, в пировиноградной кислоте, содержатся обе эти группы, называется «кетокислотой».
В организме кетокислоты практически неизбежно подвергаются химической реакции, в ходе которой теряют один атом углерода. Опустив подробности, скажем лишь, что в результате пировиноградная кислота (с тремя атомами углерода) превращается в уксусную (с двумя атомами углерода), как показано на рис. 59.
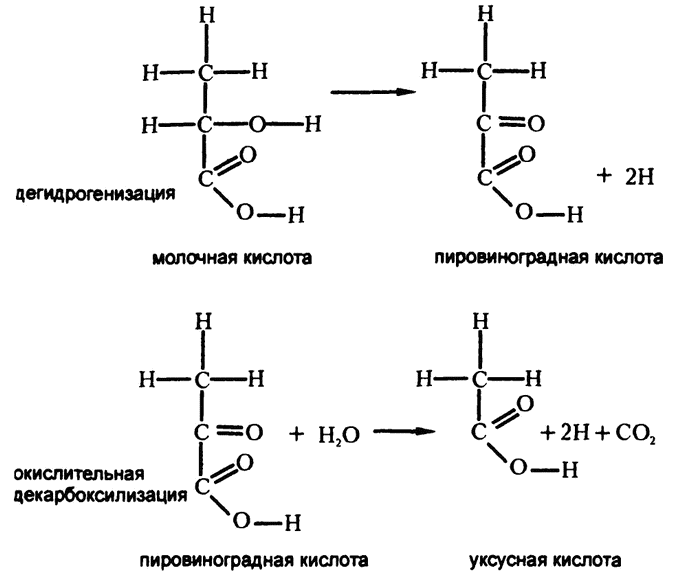
Рис. 59. Дегидрогенизация и окислительная декарбоксилизация
Такая реакция является примером «окислительной декарбоксилизации». Окислительной – потому что при ней удаляется два атома водорода, а декарбоксилизации – потому что углекислота при этом тоже удаляется. Конкретно эта реакция окислительной декарбоксилизации катализируется ферментом, в котором используется кофермент, содержащий группу атомов, представляющую собой довольно сложную молекулу «тиамин». Это и есть тот самый витамин В1? отсутствие которого вызывает у человека болезнь берибери. Именно дефицит этого витамина Эйкман изучал в 90‑х годах XIX века (см. главу 18), и именно связанные с ним исследования положили начало изучению витаминов в целом. И вот пример того, для чего нужен витамин, – без него реакция превращения пировиноградной кислоты в уксусную не состоится и вся метаболическая цепочка реакций начнет пробуксовывать.
При переводе органических веществ в воду и углекислый газ вода образуется в ходе реакций дегидрогенизации, поскольку атомы водорода, теряемые такими веществами, как молочная кислота, в итоге объединяются с кислородом. Углекислый газ формируется по большей части в ходе реакций окислительной декарбоксилизации кетокислот.
Именно на этапе дегидрогенизации и производится используемая организмом энергия. Само по себе устранение из вещества углекислоты не позволяет получить достаточно энергии для формирования высокоэнергетических фосфатных связей. Значит, можно сделать вывод, что организм получает энергию за счет сжигания водорода, а сжигание углерода – в общем‑то случайное побочное явление. Это неудивительно, поскольку, как я уже писал (см. главу 8), при сжигании водорода высвобождается гораздо больше тепла, чем при сжигании углерода.
Следующим этапом уксусная кислота добавляется в щавелево‑уксусную и попадает в цикл Кребса (как вы помните, щавелево‑уксусная кислота оказалась одним из первых веществ, участие которого в цикле стало известно).
В начале 40‑х годов XX века подробности процесса добавления были еще неизвестны. Однако уже в 1947 году Липман (первооткрыватель высокоэнергетических фосфатных связей) сумел выделить вещество, служащее коферментом для данной реакции, и назвал его «кофермент А».
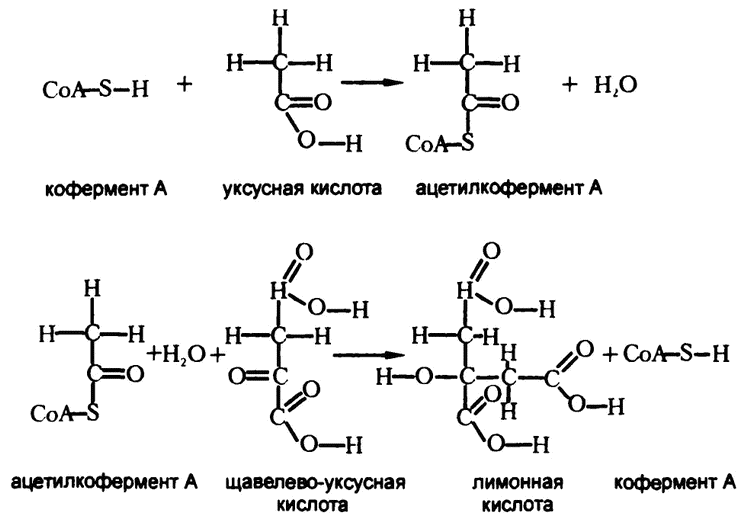
Рис. 60. Кофермент А
Молекула кофермента А достаточно сложна, и я не буду вас мучить. Большая часть ее – это пантотеновая кислота, витамин В3, который организм сам производить не умеет, а значит – должен получать с пищей.
Та часть кофермента А, которая участвует в реакции, – это тиоловая группа (– S – Н). Поэтому молекулу кофермента А часто обозначают так:
CoA – S – Н.
Кофермент А конденсируется с уксусной кислотой, как показано на рис. 60, образуя ацетилкофермент А. На самом деле он участвует в реакции окислительной декарбоксилизации пировиноградной кислоты, так что сама уксусная кислота здесь образоваться не может. Сразу получается ацетилкофермент А.
Ту часть молекулы уксусной кислоты, которая присоединена к коферменту А, иногда называют «двухуглеродным фрагментом». Это дань традиции, происходящей из тех времен, когда точное строение этой части еще было неизвестно.
В организме происходит много реакций, при которых двухуглеродный фрагмент переходит от одного вещества к другому, и в каждом случае именно кофермент А является той рабочей лошадкой, которая выполняет эту задачу. Позже было установлено, что кофермент А участвует и в реакции с альфа‑кетоглютаровой кислотой, тоже задействованной в цикле Кребса, и там выполняет задачу переноса уже четырехуглеродного фрагмента. Не стоит считать, что кофермент А может иметь дело только с двухуглеродными фрагментами, и мы еще вернемся к этой теме, когда речь зайдет о других его задачах по переносу.
Добавление к щавелево‑уксусной кислоте ацетилкофермента А показано и на рис. 60. Эта сложная реакция (но при тщательном рассмотрении вы сами убедитесь, что каждому атому слева соответствует атом справа), в результате которой образуется лимонная кислота со своими шестью атомами углерода, а кофермент А формируется заново и отправляется за следующей молекулой уксусной кислоты.
После образования лимонной кислоты она проходит через ряд реакций и в итоге избавляется‑таки от добавленного к ней двухуглеродного фрагмента. Таким образом, лимонная кислота превращается обратно в щавелево‑уксусную. Теперь щавелево‑уксусная кислота снова может приобрести двухуглеродный фрагмент, который снова будет утерян при следующей итерации цикла Кребса, и так далее.
Формулы, которые я приводил в данной главе, иллюстрируют основные этапы процесса: окислительную декарбоксилизацию, дегидрогенизацию, катализируемый перенос коферментами и так далее. Так что теперь мы можем перейти, уже без формул, к схеме‑иллюстрации процесса катаболизма молочной кислоты. Эта схема приведена на рис. 61, и, уверяю вас, там показаны только самые основные моменты.
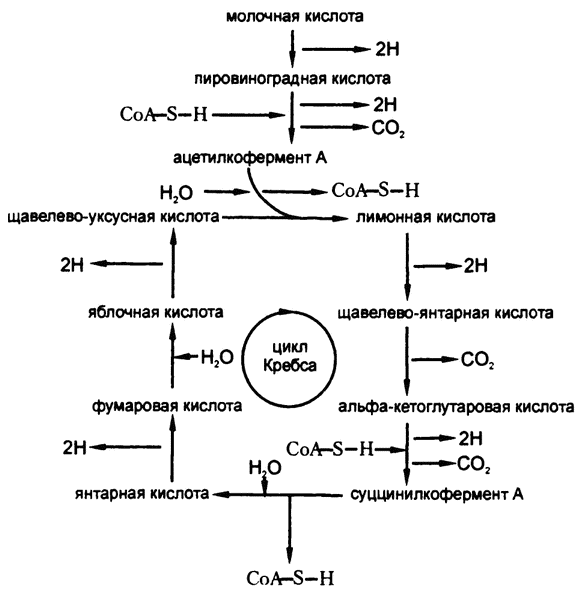
Рис. 61. Катаболизм молочной кислоты
Как видите, молочная кислота сначала подвергается одной дегидрогенизации, превращаясь таким образом в пировиноградную, затем – второй в процессе образования ацетилкофермента А, а затем – еще четырем в процесс цикла Кребса. Всего получается шесть дегидрогенизаций, в ходе которых теряется двенадцать атомов водорода.
Но ведь в молекуле молочной кислоты всего шесть атомов водорода, как вы сами можете убедиться, взглянув на формулу, изображенную на рис. 59. Как же она ухитряется потерять двенадцать?
Как показывает общая схема цикла Кребса, на входе в него на трех разных участках поступают три молекулы воды. Они‑то и предоставляют те самые недостающие шесть атомов водорода.
Кроме того, серия изменений, которым подвергается молочная кислота и вещества, в которые она последовательно превращается, включает в себя де‑карбоксилизацию в процессе образования ацетил‑кофермента А и еще две декарбоксилизации в ходе цикла Кребса для образования всего трех молекул углекислоты.
Соответственно, общее выражение будет выглядеть так:
С3H6O3 + 3H2O → 3CO2 + 12H.
молочная кислота
Возникает несколько вопросов. Во‑первых, давайте вернемся к судьбе наших атомов водорода. Понятно, что должен использоваться кислород, поскольку, в конце концов, и сам цикл Кребса был открыт только благодаря исследованиям процессов потребления кислорода. Однако кислород ведь не принимает атомы водорода напрямую, поскольку эксперименты показывают, что перекись водорода не образуется в тканях, даже если ткани бедны каталазой.
Значит, кислород принимает атомы водорода через какое‑то посредничество, так чтобы в итоге получалась не перекись водорода, а вода. Остается предположить, что атомы водорода сначала принимает какая‑то другая молекула, не кислород, а затем – передает кислороду. Так же как кофермент А переносит двухуглеродные фрагменты, какой‑то другой кофермент должен переносить и атомы водорода.
На самом деле такие «переносчики водорода» были открыты еще за поколение до того, как был обнаружен кофермент А. Первым из переносчиков водорода был вообще самый первый из открытых коферментов – козимаза, обнаруженная Харденом и Янгом (см. главу 18). В начале 30‑х годов XX века была установлена и молекулярная структура козимазы. Я не буду сейчас углубляться в подробности, как и в отношении кофермента А, но основные моменты перечислю.
Прежде всего, в состав козимазы входит молекула сахара из пяти атомов углерода, две фосфатные группы и азотсодержащее атомное кольцо. Все это позволяет отнести молекулу к классу нуклеотидов. Азотсодержащее кольцо имеет пиридиновую форму, как я уже упоминал в главе 18, так что вся молекула получила научное название «дифосфопиридиннуклеотид», или, в сокращенной записи, ДПН. В 1934 году Варбург, изобретатель респирометра, выделил очень похожий кофермент, который отличался от козимазы только тем, что в его состав входят три, а не две фосфатные группы. Соответственно, это вещество получило название «трифосфопиридиннуклеотид», в сокращении ТПН.
И в ДПН, и в ТПН входит особый вариант пиридинового кольца, известный как никотинамид, который организм может образовывать только на основе похожего вещества – никотиновой кислоты. Значит, либо никотинамид, либо никотиновая кислота тоже должны присутствовать в пище – это тоже витамины группы В.
На протяжении 30‑х и 40‑х годов XX века ученые выделяли все больше и больше ферментов, катализирующих дегидрогенизацию различных веществ, и все открытые ферменты получили общее название «дегидрогеназы». В состав большой части дегидрогеназ входят в качестве необходимого кофермента либо ДПН, либо ТПН. Поэтому дегидрогеназы получили еще одно название, свидетельствующее о важности для их строения пиридинового кольца: «пиридин‑ферменты».
Активной группой, «режущей кромкой», так сказать, каждого пиридин‑фермента является именно ДПН или ТПН. Однако специфику фермента определяет все же аминокислотная составляющая (апофермент). Именно она представляет собой решающий фактор, благодаря которому вступить в контакт с коферментом, а значит – послужить субстратом, могут только особые вещества. То есть десяток разных пиридин‑ферментов, в состав которых входят десять различающихся между собой аминокислот, будут катализировать дегидрогенизацию десяти разных веществ, и ни один из ферментов не сможет повлиять на девять «соседских» субстратов.
Вообще говоря, из шести процессов дегидрогенизации, включенных в катаболизм молочной кислоты, пять проходят с помощью пиридин‑ферментов, причем каждый раз разных. Шестая же дегидрогенизация проходит с участием янтарной кислоты, и об этом чуть позже.
Функция ДПН и ТПН – принимать атом водорода, отдаваемый субстратом. Субстрат отдает атом водорода и окисляется, а ДПН или ТПН – принимает и восстанавливается. Восстановленный ДПН или ТПН можно обозначить, не вдаваясь в тонкости строения, просто как ДПН‑ 2Н или ТПН‑ 2Н. Таким образом, дегидрогенизацию молочной кислоты в пировиноградную, катализируемую пиридин‑ферментом «дегидрогеназой молочной кислоты», можно представить, не вдаваясь в подробности (поскольку структурные формулы молочной и пировиноградной кислот вы можете посмотреть на рис. 59), так:
С3Н6О3 + ДПН → С3Н4О3 + ДПН ∙ 2Н
молочная кислота пировиноградная кислота
ДПН∙2Н далее передаст оба атома водорода другому веществу и будет снова окислен до состояния обычного ДПН. Теперь он готов снова принять два атома водорода, чтобы опять передать их дальше, и так далее.
Так что же это за «другое вещество», которому ДПН отдаст водород? Кислород?
Давайте посмотрим. Если пиридин‑фермент принимается за работу по дегидрогенизации субстрата, например, молочной кислоты, при отсутствии других ферментов и субстратов и в азотистой атмосфере, то реакции не произойдет. Да, атомы водорода ДПН примет, но передать их будет нечему. А общее количество присутствующих в системе молекул ДПН настолько мало по сравнению с количеством молекул молочной кислоты, что даже полное восстановление всего запаса ДПН не отразится сколь‑нибудь заметным окислением молочной кислоты. Пока ДПН не сможет отдавать получаемые атомы водорода, реакция не запустится.
Исправить положение удается, если добавить в систему кислород, из чего становится ясно, что кислород принимает водород в реакциях с участием пиридин‑фермента.
Однако, если добавить в систему другие вещества, например метиленовую синь, то реакция состоится и в отсутствие кислорода. Метиленовая синь будет принимать атомы водорода у ДПН ∙ 2Н. Это вещество, как понятно из названия, имеет синий цвет, но восстановленная метиленовая синь – бесцветна. Так что за ходом реакции можно следить по постепенному обесцвечиванию метиленовой сини.
Это, конечно, интересно, но практического решения проблемы нам не дает, поскольку в живой ткани метиленовая синь не встречается. Эти экспериментальные данные свидетельствуют лишь о том, что ДПН и ТПН могут при определенных обстоятельствах отдавать атомы водорода другим веществам, но каким же именно веществам они отдают атомы в живой ткани?
Отвлечемся ненадолго, а потом продолжим.
В 30‑х годах XX века (а то и раньше) ученые нередко выделяли из тканей желтое вещество, получившее название «флавин», от латинского «желтый». К концу десятилетия было установлено строение этих веществ – обнаружилось, что они содержат систему из трех колец, которую организм сам создать не в силах и, опять же, должен получать с пищей в виде «рибофлавина» – одной из разновидностей витамина В.
Варбург показал, что флавины связаны с системами ферментов, которым и передают свой желтый цвет, и первый обнаруженный фермент такого рода получил название «старый желтый фермент Варбурга». В составе флавинов были в конечном итоге найдены два важных кофермента – «флавинмононуклеотид» (ФМН) и «флавинадениндинуклеотид» (ФАД).
Ферменты, в состав которых входят ФМН или ФАД, называются «флавоферментами», и, как оказалось, они, как и ферменты, включающие в себя ДПН или ТПН, катализируют дегидрогенизацию. К примеру, дегидрогенизация янтарной кислоты (единственная, как я уже говорил, во всем цикле катаболизма молочной кислоты, обходящаяся без пидидин‑фермента) происходит именно под каталитическим действием «дегидрогеназы янтарной кислоты», коферментом которой является ФАД.
В этом случае, как и в предыдущем, предназначение коферментов – также принимать из субстрата атомы водорода, становясь при этом ФМН‑ 2Н или ФАД‑ 2Н, а затем отдавать этот водород чему‑то еще.
Но есть и разница. Коферменты флавинов могут отдавать атомы водорода кислороду с образованием перекиси водорода. Вследствие этого дегидрогеназы флавоферментов называют еще «аэробными дегидрогеназами», поскольку они могут осуществлять каталитическую деятельность в присутствии кислорода, как единственного принимающего водород вещества. А дегидрогеназы пиридин‑ферментного класса называют «анаэробными дегидрогеназами», потому что они не умеют этого делать.
Таким образом, если в систему, содержащую пиридин‑ферменты, добавить флавофермент и субстрат (которые сами по себе в реакцию не вступают), то флавофермент примет атом водорода у пиридин‑фермента и передаст его кислороду. Сам по себе флавофермент в прямой контакт с субстратом не вступает, а пиридин‑фермент – не вступает в контакт с кислородом. Однако вместе они успешно катализируют дегидрогенизацию субстрата и восстановление кислорода в перекись водорода, как схематически изображено на рис. 62.
Перенос проходит, как видите, в три этапа, на каждом из которых происходит своя окислительно‑восстановительная реакция. Как я уже объяснял в конце предыдущей главы, атомы водорода постепенно движутся по пути увеличения окислительного потенциала.
Почему же атомы водорода не могут сразу от ДПН∙2Н переходить к кислороду? Такой процесс тоже подразумевал бы увеличение окислительного потенциала. Зачем же нужен промежуточный процесс? Тут можно воспользоваться следующей аллегорией: если сила тяжести тянет человека вниз, он может, подчиняясь ей, сделать два шага по ведущим вниз ступенькам, а может и один – через ступеньку. Как правило, необходимость в промежуточном восстановленные формы окисленные формы
этапе обусловливается понижением энергии активации. Видимо, организм не может предоставить сразу достаточное количество энергии для того, чтобы преодолеть порог, необходимый для прямой передачи атомов водорода от органического вещества к кислороду. Так что приходится пользоваться промежуточным этапом в виде неорганического посредника.
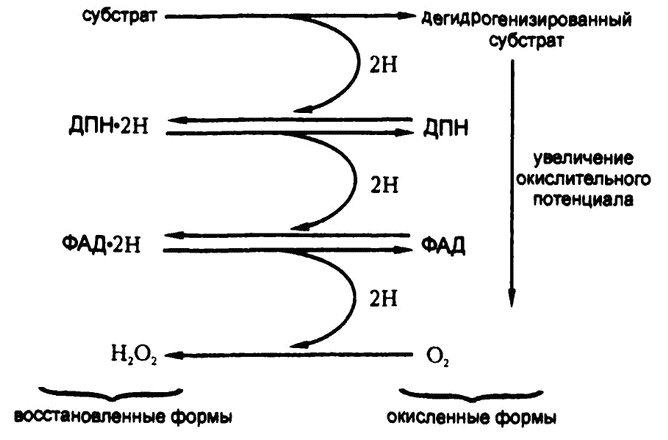
Рис. 62. Этапы дегидрогенизации
В 50‑х годах XX века было обнаружено, что в флавоферментах содержатся атомы металла, а в пиридин‑ферментах – нет. То есть, например, дегидрогеназа янтарной кислоты имеет в своем составе атомы железа, другие флавоферменты – атомы меди или молибдена. Железо может принимать форму как двух‑, так и трехвалентного иона (Fe2+ и Fe3+). Приняв один электрон, трехвалентный ион железа превращается в двухвалентный, а тот, потеряв электрон, – обратно в трехвалентный. Так, переключаясь из одной формы в другую, атом железа может выступать переносчиком электронов. Такую же работу может выполнять и медь, переключаясь из состояния одновалентного иона меди (Cu+) в состояние двухвалентного (Cu2+) и обратно. Молибден тоже обладает подобным свойством.
Такой перенос электронов могут осуществлять только металлосодержащие флавоферменты, а чисто органические пиридин‑ферменты – не могут. И видимо, перенос электронов – необходимая часть процедуры снижения энергии активации переноса водорода к молекуле кислорода. Ничего точнее сказать, увы, по сей день невозможно.
Можно подумать, что теперь ситуация с атомами водорода, полученными в ходе катаболизма, благополучно прояснена, но это, конечно, не так. Когда кислород принимает атомы водорода из флавоферментов, образуется перекись водорода. Однако в целом нам известно, что перекись водорода в организме не образуется, так что приведенная схема явно не завершена. Должно быть что‑то еще.
В 1925 году британский биохимик Д. Кейлин занимался изучением того, как размешанные в растворе взвеси истолченных тканей различных видов – от бактериальных до нервных тканей высших животных – поглощают свет. Он обнаружил около полудюжины разных полос поглощения и приписал их наличие предположительному существованию вещества, которое назвал «цитохром» (от греческих слов, означающих «клетка» и «цвет»). Дальнейшие исследования показали, что полосы поглощения существуют попарно, и каждая пара полос поглощения привязана к своему веществу. Естественно, они получили названия «цитохром a», «цитохром b» и «цитохром с». Со временем выяснилось, что даже такое деление не совсем точно, было вещество близкое к цитохрому a, но все же не полностью с ним идентичное, и оно получило название «цитохром a3».
Единственным цитохромом, который удалось достаточно легко выделить из взвеси тканей, оказался цитохром с. Обнаружилось, что это сравнительно простой белок, молекулярный вес его – около 13 000, и каждая молекула этого вещества содержит один атом железа. Этот атом является частью гема, такого же гема, какой входит и в состав гемоглобина (см. главу 18). Дальнейшие исследования показали, что и в других цитохромах тоже содержится железо – и в каждом случае оно входило в состав либо гема, либо очень похожей на гем группы атомов.
Соответственно, цитохромы получили видовое название «гемоферменты». Каталаза, как я уже упоминал в предыдущей главе, тоже является примером гемофермента, но она не выполняет функций цитохромов. Гемоглобин – это гемосодержащий белок, но не гемофермент. Не все ферменты, в составе которых имеется железо, – гемоферменты. К примеру, в состав дегидрогеназы янтарной кислоты тоже входит атом железа, но не как часть гемовой группы.
Когда была установлена схема передачи водорода («дыхательная цепочка»), вскоре стало ясно, что цитохромы должны быть в нее где‑то включены. Они присутствуют практически во всех клетках, за исключением разве что клеток некоторых «обязательно анаэробных» бактерий – то есть таких, которые могут жить только при отсутствии кислорода. Факт того, что в их клетках цитохромы отсутствуют в сочетании с тем, что эти бактерии не могут использовать кислород, сам по себе уже свидетельствует в пользу важности роли цитохромов в дыхательных цепочках.
И опять же, любое вещество, препятствующее деятельности цитохромов – а особенно колебаниям атома железа между двух‑ и трехвалентным состоянием, что прекращает передачу электрона, – препятствует и поглощению кислорода. Воздействие цианидных групп (–С=N) таких веществ, как синильная кислота (HCN) или цианистый калий (KCN), приводит к прочному застыванию атома железа в одном из состояний, в результате чего дыхание быстро и навсегда прекращается (именно поэтому цианиды так ядовиты).
О положении цитохромов в цепочке можно судить по окислительным потенциалам, приведенным в таблице 9. Очевидно, цитохромы стоят где‑то после флавинов, принимают электроны от атомов водорода и передают их по одному путем колебания железа между двух‑ и трехвалентным ионным состоянием, от «b» к «c», от «c» к «a», от «a» к «a3».
Возможно, при этом передаются и сами атомы водорода, хотя в этом отношении точных данных нет.
Однако где‑то ведь цепочка должна закончиться! На каком‑то этапе водород должен быть передан кислороду – и это этап цитохрома a3. Окислительный потенциал системы «кислород/вода» равен +0,80, и он в конце концов берет свое. Поскольку цитохром a3 может использовать кислород в качестве получателя водорода, в результате чего образуется вода, а не перекись водорода (получается, вот он, фермент, катализирующий разрыв связи О–О!), то его можно назвать «оксидазой». На самом деле именно под этим названием цитохром a3 и известен по большей части – «цитохромоксидаза».
Все, получили воду, можно теперь расслабиться. Теперь дыхательная цепочка собрана до конца.
Опять возникает вопрос: почему же она оказывается такой длинной? Почему же требуется так много коферментов и простетических групп, чтобы перенести атомы водорода от субстрата к ферменту?
Таблица 9
Дата добавления: 2016-01-26; просмотров: 3499;