В. Активация перекисного окисления липидов
Хаотропный эффект избытка жирных кислот и лизофосфатидов поддерживает активацию перекисного окисления липидов (ПОЛ), инициируемого накоплением в гипоксической клетке активных форм кислорода (АФК). Генерация последних связана с Са2+ - зависимым повреждением митохондрий и формированием избытка доноров электронов – восстановленных кофакторов.
Образование активных (токсичных) форм кислорода (в невозбужденном состоянии кислород нетоксичен) связано с особенностями его молекулярной структуры: О2 содержит два неспаренных электрона с параллельными спинами, которые не могут образовывать термодинамически стабильную пару и располагаются на разных орбиталях. Каждая из этих орбиталей может принять еще один электрон. Таким образом, полное восстановление молекулы кислорода происходит в результате четырех одноэлектронных переносов:
 е- е- е- е-, Н+
е- е- е- е-, Н+
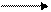 О2 О2- Н2О2 `ОН + Н2О 2Н2О
О2 О2- Н2О2 `ОН + Н2О 2Н2О
Образующиеся в ходе неполного восстановления молекул кислорода супероксид (О2-), пероксид (Н2О2) и гидроксильный радикал (`ОН) – активные формы кислорода, являются окислителями, что представляет серьезную опасность для многих структурных компонентов клетки (Авдеева Л.В., Павлова Н.А., Рубцова Г.В., 2005). Особенно активен гидроксильный радикал (•OH), взаимодействующий с большинством органических молекул. Он отнимает у них электрон и инициирует таким образом цепные реакции окисления.
Основной путь образования АФК в большинстве клеток – утечка электронов из цепи их передачи (дыхательной цепи) и непосредственное взаимодействие этих электронов с кислородом (Губарева Л.Е., 2005). В качестве еще двух источников могут выступать реакции с участиемоксидаз, использующих молекулярный кислород как акцептор электронов и восстанавливающих его до Н2О или Н2О2 и реакции с участиемоксигеназ, включающих один (монооксигеназы) или два (диоксигеназы) атома кислорода в образующийся продукт реакции. В условиях дефицита в тканях кислорода, т.е. в ситуации, когда «спрос» (восстановленные кофакторы) превышает «предложение» (количество молекул кислорода), вероятность усиления образования АФК резко возрастает. Инициируемые ими свободнорадикальные реакции, приводят к повреждению клеточных и субклеточных структур, включая митохондрии, молекулы ДНК и белка. И хотя вклад АФК в развитие гипоксического некробиоза (в отличие от реперфузионного синдрома) расценивается в качестве доминирующего механизма не всеми авторами (Зайчик А.Ш., Чурилов Л.П., 1999), тем не менее их участие в активации свободно-радикальных процессов в клетке, включая ПОЛ, является решающим.
Следует отметить, что ПОЛ представляя собой саморазвивающуюся цепную реакцию, постоянно протекает в клетке, играя роль необходимого звена в ее жизнедеятельности и в адаптационных реакциях. Благодаря перекисному окислению в молекуле фосфолипидов клеточных мембран, содержащих во втором положении жирную кислоту, появляются полярные гидроперекисные группировки (гидроперекиси липидов), обладающие детергентным действием. Появление таких группировок увеличивает подвижность полипептидных цепей, т.е. облегчает конформационные изменения молекул белков, что сопровождается ростом активности мембраносвязанных ферментов, к которым по существу относятся все ферментные системы клетки. И лишь чрезмерная активация ПОЛ, затрагивающая более 3-5% фосфолипидов мембран, превращает его из регуляторного механизма в звено патогенеза их повреждения при клеточной гибели (Ю.А. Владимиров, 1987; 2000).
В результате активации ПОЛ, инициируемого АФК, и прежде всего – гидроксильным радикалом (•OH), происходит образование новых вторичных радикалов: липидного (L•), алкоксильного (LO•), перекисного (LOO•). Рис. 28.
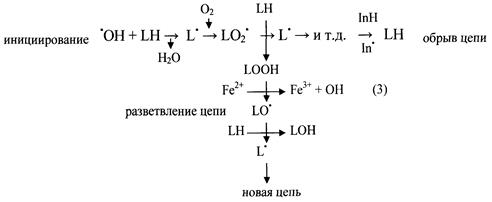
Рис. 28. Перекисное окисление липидов и образование вторичных радикалов
(Ю.А. Владимиров, 2001)
Химическая активность этих вторичных органических радикалов ниже, чем у гидроксильного радикала (•OH), но они активно вовлекаются в цепную реакцию ПОЛ, поддерживая и усугубляя повреждения липидного бислоя клеточных мембран.
Модифицирующие эффекты ПОЛ в отношении фосфолипидов определяют цепь дальнейших событий (Архипенко Ю.В. с соавт., 1983; Меерсон Ф.З., 1989; Владимиров Ю.А., 2001). Прежде всего, в молекулах фосфолипидов, содержащих во втором положении жирную кислоту, появляется полярная гидроперекисная группировка (рис. 29).
При этом накопление гидроперекисей липидов сопровождается уменьшением количества ненасыщенных липидов. При умеренной активации ПОЛ, как отмечалось выше, появление в микроокружении интегральных белков полярных продуктов ПОЛ, обладающих детергентным действием, вызывает увеличение подвижности полипептидной цепи, что, как правило, сопровождается увеличением каталитической активности ферментов. При избыточной активацииПОЛ главное значение приобретает уменьшение количества непредельных фосфолипидов.
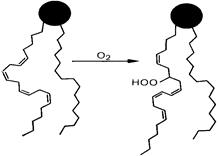
Рис. 29. Образование гидроперекиси фосфолипида, начальный этап процесса ПОЛ
(Ф.З. Меерсон, 1984).
· Значительное уменьшение содержания непредельных фосфолипидов в мембране под влиянием ПОЛ, повышает регидность (микровязкость) ее липидного бислоя, что сопровождается снижением конформационной подвижности полипептидных цепей белков, встроенных в мембрану (эффект «вмораживания»). Поскольку такая подвижность необходима для нормального функционирования ферментов, рецепторов и каналоформеров, их функциональный ответ ингибируется (рис. 30).
Рис. 30 Изменение активности Са-АТФазы в мембранах саркоплазматического
ретикулума в результате модификации липидного окружения этого фермента
процессом ПОЛ(Ф.З. Меерсон, 1984)
А - исходное состояние; Б - умеренная активация Са-АТФазы; В - ингибирование-Са-АТФазы.
· Окисленные в ходе активации ПОЛ фосфолипиды подвергаются латеральной диффузии вдоль мембраны и образуют ассоциаты (кластеры), фиксированные взаимодействием фосфолипидов между собой и молекулами воды. Эти участки мембраны приобретают гидрофильность. Располагаясь друг против друга в каждом из монослоев липидного бислоя, такие ассоциаты образуют каналы в мембране, увеличивая ее проницаемость для воды, кальция и других ионов (рис. 31).
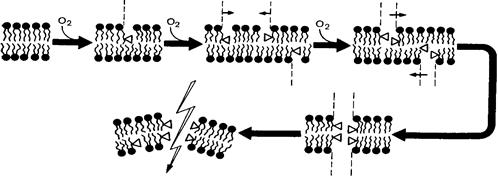
Рис. 31.Схема образования перекисных кластеров и фрагментация мембраны при индукции перекисного окисления липидов(Ф.З. Меерсон, 1984)
Светлый треугольник — гидроперекисная группа.
· Образующиеся продукты распада гидроперекисей фосфолипидов (малоновый, глутаровый и др. диальдегиды) взаимодействуют со свободными аминогруппами мембранных белков, образуя межмолекулярные сшивки и инактивируя эти белки (рис. 32). In vivo этот процесс приводит к образованию т.н. оснований Шиффа пигмента изнашивания липофусцина.
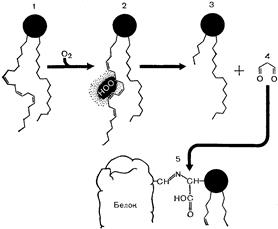
Рис. 32. Образование сшивок и ингибирование мембранных белков-ферментов в результате активации ПОЛ(Ф.З. Меерсон, 1984)
Последний представляет собой смесь липидов и белков, связанных между собой поперечными ковалентными связями и денатурированными в результате взаимодействия с химически активными группами (диальдегидами) продуктов ПОЛ. Этот пигмент фагоцитируется, но не гидролизуется ферментами лизосом, и поэтому накапливается в клетках в виде пигментных пятен, особенно на дорзальной поверхности ладоней у пожилых людей.
Гидроперекись (2), образовавшаяся в результате реакции фосфолипидов (1) с молекулярным кислородом, распадается на фосфолипид с укороченной углеводородной цепью во втором положении, сходный с лизофосфолипидами (3) и короткий углеводородный фрагмент – диальдегид (4). Взаимодействие бифункциональной по своей природе молекулы диальдегида с аминогруппами одновременно двух молекул белков приводит к формированию сшивки (5).
· Под влиянием ПОЛ происходит окисление сульфгидрильных (-SH) групп мембранных белков: ферментов, ионных каналов и насосов, что приводит к падению их активности.
· Образование полярных продуктов окисления способствует возрастанию на мембране отрицательного поверхностного заряда, обусловливающего фиксацию на ней полиэлектролитов. Среди последних – некоторые белки и пептиды, формирующие белковые поры – один из факторов снижения электрической стабильности мембран.
· Увеличение полярности внутренней оболочки мембраны обусловливает проникновение воды в липидный бислой – т.н. «водную коррозию мембраны».
· «Выталкивание» из мембраны части окислившихся полиненасыщенных жирных кислот приводит к уменьшению площади ее липидного бислоя.
Таким образом, на этом этапе развития гипоксического повреждения клеток ключевым звеном патогенеза выступает дезорганизация липидного бислоя мембран, осуществляемая при участии ионов кальция и липидной триады: активации липаз и фосфолипаз; детергентного действия избытка жирных кислот и лизофосфолипидов, а также активации перекисного окисления липидов.
Существенный вклад в эту дезорганизацию вносят также: механическое (осмотическое) растяжение мембран и адсорбция на липидном бислое полиэлектролитов, способствующие увеличению их порозности. В совокупности указанные нарушения обусловливают снижение электрической прочности мембран и возникновение электрического пробоя липидного бислоя собственным мембранным потенциалом(рис. 33). Последний рассматривается как терминальный механизм нарушения барьерной функции мембраны (Владимиров Ю.А., 2001).
Этот этап патогенетической цепи повреждения клеток при гипоксии, характеризующийся нарастающей утратой барьерной и матричной функций мембран, определяет переход обратимых изменений в клетке – в необратимые.
Последующее развитие событий связано с формированием повреждений клеточных структур, непосредственно приводящих к клеточной гибели. Существенно, что механизмы этих повреждающих эффектов также тесно связаны с повышенным содержанием в цитозоле ионов Са2+.
Патогенетические последствия избытка ионов кальция в заключительной стадии гипоксического повреждения клеток (стадия некробиоза) не ограничиваются активацией липаз и фосфолипаз. Ионы Са2+ прямо участвуют в прямых эффектах повреждения клеточных структур и апоптотической гибели клеток. К числу этих эффектов относятся:
· Разрушение цитоскелета,которое связано с Са2+-зависимой активацией кальпаинов. Происходит деструкция некоторых белков цитоплазмы (β-актин, фодрин), что вызывает деформацию клеток, ограничивающую возможность их взаимодействия с микроокружением, а также способность к восприятию регуляторных сигналов. Слабость цитоскелета способствует дезинтеграции некоторых надмолекулярных комплексов в клетке, в частности, отсоединению рибосом от мембран шероховатого эндоплазматического ретикулума. В результате происходит насыщение цитоплазмы белковыми молекулами, подвергающихся деградации.
· Механическое повреждение клеточных структур,обусловленное Са2+ активацией сократительной функции миофибрилл с одновременной утратой ими способности к расслаблению. Такие контрактурные сокращения сопровождаются механическим повреждением сократительных структур клетки.
· Омыление и эндогенный детергентный эффект.Накопление в клетке жирных кислот в присутствии избытка ионов Са2+ (и Na+) приводит к образованию мыл – солей высших жирных кислот. По этой причине гидролиз сложноэфирных связей называется омылением. Образование мыл в цитозоле резко увеличивает его детергентную активность которая в буквальном смысле растворяет липидные мембраны (Зайчик А.Ш., Чурилов Л.П., 1999). Мыла, разрушая мембраны органоидов, обрушивают на клетку удар гидролаз, активных радикалов и других метаболитов, которые до этого момента были изолированы в различных отсеках клетки. Этот эндогенный эффект имеет решающее значение в формировании финальной стадии клеточной гибели.
· Наряду с участием в некробиозе, ионы кальция участвуют в реализации механизмов апоптотической гибели клеток. Среди последних: повышение активности Са2+-зависимых эндонуклеаз и кальпаинов. Подобная активация несет в себе угрозу для клетки, инициируя ее апоптотическую гибель либо вследствие фрагментации ДНК (эндонуклеазами), либо в результате протеолиза антиапоптотических белков (bcl-2) кальпаинами. Апоптозу может способствовать и кальпаининдуцированная деградация протеинкиназы С(ПКС), реализующую, в основном, антиапоптотические эффекты и повышающую устойчивость клеток к токсическим продуктам обмена.
· Более того, избыток ионов Са2+ сам способствует образованию токсических продуктов, в роли которых могут, в частности, выступать молекулы оксида азота в высоких концентрациях, создаваемых Са2+-активацией индуцибельной NO-синтазы. Наиболее ярко такой эффект проявляется при т.н. глутаматной гибели нейронов, возникающей при гипоксии (ишемии мозга). Инициация развития событий в этом случае связана с дефицитом энергии в нейронах, выходом ионов калия, деполяризацией мембран и повышением внутриклеточного пула Са2+ в результате длительного открытия потенциал зависимых кальциевых каналов (рис. 34).

Рис. 34. Механизм развития глутаматной гибели нейронов при гипоксии
Следствием избытка ионов кальция в цитоплазме является повышенное выделение нейромедиатора (глутамата) глутаматергическими нейронами в синаптическую щель. Восприятие данного сигнала постсинаптическими нейронами осуществляется с помощью НМДА-рецепторов (наиболее хорошо изученный подтип рецепторов глутамата с высоким сродством к синтетической аминокислоте Н-метил-Д-аспартату), чувствительность которых к медиатору в условиях гипоксии значительно возрастает (Крыжановский Г.Н., 1997). Результатом «глутаматной бомбардировки» (Акмаев И.Г., 1996; Акмаев И.Г., Гриневич В.В., 2001) постсинаптического нейрона является открытие в нем ионных каналов, приводящее к увеличению поступления кальция в клетку и активация нейрональной NO-синтазы (NOS). Продуцируемый под ее влиянием оксид азота, имея малый размер и липофильную природу молекулы, диффундирует во внеклеточное пространство и поступает через мембраны в близлежащие клетки (нейроны), оказывая на них токсическое влияние. Основу этого токсического влияния составляет энергетический дефицит клеток. Механизм формирования такого дефицита связан со способностью NO вызывать S-нитрозилирование клеточных железосодержащих белков(аконитаза ЦТК, комплексы I-III цепи переноса электронов в МТХ) и их инактивацию. Кроме того, под влиянием NO происходит рибозилирование и нитрозилированиеглицеральдегид-3-фосфатдегидро-геназы, обусловливающей торможение гликолиза. Наконец, при взаимодействии NO с другим радикалом – О2- образуется пероксинитрит-анион (ONOO-), вызывающий необратимое ингибирование железосодержащих белков.
За счет образования ONOO- возможно включение апоптотического механизма гибели клеток путем реализации следующего каскада:

Особенностью глутаматной гибели нейронов является отсутствие гибели самих NO-продуцирующих нейронов, оказывающихся защищенными от токсического действия NO. Механизм этой защиты связывают с активацией супероксиддисмутазы (СОД) и (или) с переходом NO в окисленную форму (NO+). По сути здесь прослеживается прямая аналогия с макрофагами, которые, продуцируя NO, сами проявляют к нему устойчивость.
Таким образом, гибель клетки при гипоксии представляет собой закономерное развертывание цепи событий, включающих формирование энергодефицита, ингибирование основных метаболических путей, активацию липидной триады и последующее необратимое повреждение клеточных структур. Центральным звеном патогенеза этих событий является повышение внутриклеточной концентрации ионов кальция, а главной мишенью – клеточные мембраны и, прежде всего – митохондрии.
Последовательность рассмотренных изменений при гипоксии (аноксии) одинакова для самых различных тканей. Об этом свидетельствуют опыты со срезами тканей, изолированными клетками и изолированными органеллами (Владимиров Ю.А., 2001). Рис. 35.
Различие состоит лишь в скорости протекания этих процессов, которая при температуре тела человека в 2-3 раза выше. Кроме того, эта скорость различна для разных тканей и с наибольшей быстротой указанные процессы протекают в ткани мозга, с меньшей – в печени, с еще более низкой скоростью – в мышечной ткани.

Рис. 35. Последовательность нарушений в клетках печени при аноксии
по Ю.А. Владимирову, 2001
XIV. ГИПЕРОКСИЯ
Гипероксия – повышенное поступление кислорода в организм. В отличие от гипоксии, гипероксия всегда носит экзогенный характер и в естественных условиях практически не встречается. В связи с этим, адаптивные механизмы к данному состоянию эффективны лишь в условиях относительно невысокой кислородной нагрузки, определяемой величиной парциального давления кислорода и продолжительностью его действия. Примером такой зависимости может служить кривая безопасных сроков дыхания кислородом человека (рис. 36).
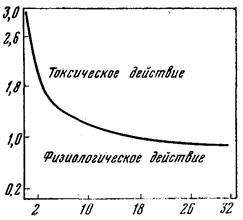
Рис. 36.Граница действия кислорода на человека(по Hartmann, 1966).
Цитируется по А.Г. Жиронкину (1979).
По оси абсцисс - длительность дыхания кислородом, часы; по оси ординат - парциальное давление кислорода, атм.
Как видно из рисунка, зона т.н. «физиологического действия кислорода» наиболее продолжительна при небольших значениях его парциального давления (около 0,5 атм.), когда защитно-приспособительные реакции в состоянии обеспечить сохранение нормального напряжения кислорода в тканях. Основу этих реакций составляют механизмы, направленные на ограничение поступления и транспорта кислорода. На это, в частности, направлена первичная реакция системы внешнего дыхания,в виде снижения легочной вентиляции и показателя минутного объема дыхания.
Данные сдвиги являются следствием прекращения в условиях повышенного поступления кислорода нормальной естественной импульсации с артериальных хеморецепторов. Вместе с тем, ограничение вентиляции не только снижает поступление кислорода в организм, но и приводит к развитию гиперкапнии. Последняя определяет вторую фазу реакции системы дыхания, характеризующуюся усилением вентиляции, направленным на снижение РаСО2 и ликвидацию газового ацидоза. Важнейшим сдвигом со стороны системы кровообращения при гипероксии является закономерное сужение мелких кровеносных сосудов, сопровождающееся ростом периферического сопротивления, замедлением общего и локального кровотока, повышением диастолического давления. Еще одним проявлением реакции со стороны этой системы служит брадикардия, регистрируемая до появления признаков кислородного отравления. Изменения со стороны системы кровив ответ на гипероксию проявляются в начальный период преходящей эритропенией и снижением уровня гемоглобина, что обусловлено перемещением тканевой жидкости в кровь и депонированием эритроцитов (Жиронкин А.Г., 1979).
При возрастании парциального давления кислорода во вдыхаемой газовой смеси, на первый план выступает его токсическое действие, поскольку защитный эффект приспособительных реакций минимизируется. В этой зоне кислород уже играет роль фактора не обеспечивающего, а угнетающего окислительные процессы в тканях. Что касается механизмов самого токсического влияния, то сегодня наиболее принятой является точка зрения R. Gershman (1964), связывающего этот механизм с образованием активных форм кислорода и с активацией свободнорадикального окисления.
В условиях перенасыщения тканей кислородом, т.е. в ситуации, когда «предложение» (избыток кислорода) превышает «спрос» (количество восстановленных кофакторов, подлежащих окислению), вероятность повышенного образования АФК возрастает. Соответственно, усиливается свободнорадикальное окисление, сопровождающееся повреждением клеточных и субклеточных структур, и, прежде всего, митохондрий.
Очевидно, что дезорганизация и повреждение митохондрий будут сопровождаться нарушением цепи транспорта электронов и окислительного фосфорилирования.Т.е. нарушениями, определяющими суть понятия «гипоксия». Соответственно, такое состояние называется гипероксической гипоксией.
Повреждение клеточных и субклеточных структур при активации свободнорадикальных процессов, приводит к развитию многочисленных нарушений специфических функций различных органов и систем. Так, ингибирование ферментов в головном мозге снижает продукцию γ-аминомасляной кислоты – важнейшего тормозного медиатора, что служит одним из механизмов развития при гипероксии судорожного синдрома кортикального генеза. Нарушение продукции сурфактанта легочным эпителием обусловливает резкое уменьшение компенсаторных резервов системы внешнего дыхания, повышая поверхностное натяжение альвеол, и способствуют появлению микроателектазов. В тяжелых случаях нарушение продукции сурфактанта может сопровождаться отеком легких. У некоторых детей первого года жизни дыхание чистым кислородом приводит к развитию респираторного дистресса – бронхопульмональной дисплазии (Маляренко Ю.Е., Пятин В.Ф., 1998). Активация свободнорадикального окисления при гипероксигенации лежит в основе формирования дефектов зрения у маленьких детей, в связи с нарушением созревания фоторецепторов.
Наряду с АФК токсическое действие кислорода опосредуется и чрезмерным напряжением некоторых защитно-приспособительных реакций. К числу таких реакций, в частности, относится длительный спазм сосудов (реакция на гипероксию). У недоношенных детей он способствует развитию ретролентальной фиброплазии (образованию фиброзной ткани за хрусталиком), приводящей к слепоте. Аналогичный спазм сосудов в легких обусловливает легочную гипертензию, расстройства микроциркуляции и повреждение легочного эпителия – нарушений, предрасполагающих к развитию воспаления.
Указанные обстоятельства заставляют ограничивать применение кислорода для лечебных целей, при которых РО2 не должен превышать 380 мм рт. ст. (Березовский В.А., 1975).
Особую чувствительность к токсическому действию избытка кислорода проявляет ткань мозга плода, которая характеризуется значительно более низким напряжением кислорода, чем церебральные структуры зрелого организма. «Этот факт не является результатом несовершенства процессов кислородного снабжения организма во внутриутробном периоде, а напротив, отражает сбалансированность этих процессов, обеспечивающих, с одной стороны, адекватную оксигенацию мозга, а с другой - защиту его от избыточного потока О2» (Рагузин А.В., 1990). Экспериментально установлено, что напряжение кислорода тканей фетального мозга является относительно стабильным параметром гомеостаза внутриутробно развивающегося организма, который мало меняется даже при значительных сдвигах кислородного режима беременных животных. Такое постоянство РО2 тканей мозга плода при сдвигах РаО2 (от 50 до 370 мм рт. ст.) материнского организма определяется механизмами, локализованными прежде всего в маточно-плацентарной области, но не системными реакциями дыхания и кровообращения. К рождению формирование механизмов стабилизации кислородного гомеостаза мозга не завершено, что служит причиной более значимого (чем у взрослых) увеличения РО2 церебральных структур новорожденных при ингаляции чистым кислородом. Подобный прирост РО2 сопровождается активацией свободнорадикального окисления в ткани мозга и развитием негативных качественных изменений параметров условно оборонительных рефлексов в зрелом возрасте (Рагузин А.В., 1990). В связи с данным положением обосновывается подход к коррекции тяжелой степени гипоксии новорожденных с использованием для ингаляции не чистого кислорода, а газовых смесей с его пониженным содержанием.
Судорожная форма кислородного отравлениявозникает при остром отравлении кислородом и известна с конца XIX столетия как симптом Бэра, впервые обнаруженный и описанный этим автором. Судороги возникают, как правило, при дыхании кислородом под давлением, превышающим 3-4 атм. и очень напоминают по своему течению эпилептические судорожные припадки.
Клинически различают три стадии этого процесса (Черешнев В.А., Юшков Б.Г., 2001):
I стадия – учащение дыхания и сердцебиения, повышение артериального давления, расширение зрачков, усиление активности с отдельными подергиваниями мышц.
II стадия – стадия судорог, похожих на эпилептические с клоническими и тоническими проявлениями.
III стадия – терминальная – ослабление судорог с расстройством дыхания, которое переходит на отдельные вдохи. Смерть наступает от паралича дыхательного центра.
Дата добавления: 2015-12-29; просмотров: 6924;