Неравновесные электрохимические процессы. Электролиз
Процесс прохождения электрического тока конечной силы через электролит является неравновесным и явления, связанные с прохождением тока, зависят от времени. При этом с течением времени состояние электрохимической системы изменяется, а параметры, характеризующие процесс, зависят от силы протекающего тока. Изучение электрохимических процессов в зависимости от силы тока, их изменений с течением времени, установление механизма составляет предмет кинетики электрохимических (или электродных) процессов (кратко – электрохимической кинетики). Электрохимическая кинетика основывается как на общих положениях химической кинетики, так и на закономерностях, характерных только для электрохимических процессов.
Электрический ток может протекать в электрохимическом элементе, состоящем из электродов и электролита, в результате замыкания этого элемента или под влиянием приложенной к системе внешней разности потенциалов. В последнем случае на границах электрод – электролит возможно выделение веществ (например, металлов или газов) из электролита на электродах, растворение вещества электрода, изменение состава электролита вблизи электрода и др. Эти явления называются электролизом.
Электролитические методы получили широкое распространение при получении металлов из солевых расплавов (алюминий, магний) и растворов (медь), газоообразного хлора и раствора щелочи электролизом растворов поваренной соли, окислении и восстановлении веществ (производство персульфата, перманганата, йодоформа, электрохлорирование бензола, электровосстановление нитробензола), нанесении покрытий и т.д.
Важно отметить, что электрохимическим путем можно проводить такие реакции, которые химическим путем при обычной температуре не идут. Самопроизвольные реакции идут с уменьшением свободной энергии, а электрохимическим путем можно проводить реакции, сопровождающиеся увеличением свободной энергии, например, разложение воды электролизом при комнатной температуре. Необходимая энергия доставляется системе извне в виде энергии электрического тока.
Так как реакция в электрохимической системе состоит из электродных реакций, процессы на каждом из электродов в неравновесных условиях отличаются от равновесных:
1. Скорость электрохимических реакций на катоде и аноде различна, т.е. сила анодного и катодного токов различна. Обычно в неравновесной электрохимической системе одна из двух возможных электродных реакций, которые соответствуют двум электродам, идет преимущественно в анодном направлении и для нее Ia > Iк, а вторая – преимущественно в катодном направлении и для нее Ia < Iк.
2. В результате преимущественного протекания реакции в одном направлении масса (а иногда и природа) электрода, а также состав раствора около него меняются по сравнению с равновесными.
3. Потенциал электрода при протекании тока в общем случае не равен равновесному электродному потенциалу и его невозможно вычислить термодинамически. Его величина зависит не только от природы системы, температуры и давления, но и от силы тока.
4. Неравновесный потенциал при достижении стационарности процесса может оказаться практически независящим от времени. Это постоянное значение потенциала электрода под током называется стационарным потенциалом.
5. Напряжение для неравновесных электрохимических систем отличается от равновесного значения ЭДС. При этом напряжение электрохимического элемента меньше, а напряжение на электрохимической ванне больше, чем ЭДС.
Так как прохождение электрического тока через электрохимическую систему связано с химическими превращениями, то между количеством прошедшего электричества и количеством прореагировавших веществ существует определенная связь. Эта связь установлена Фарадеем в первых количественных законах электрохимии – законах Фарадея.
Первый закон Фарадея. Количества веществ, превращенных при электролизе, пропорциональны количеству электричества, прошедшего через электролит. Это закон можно представить уравнением:
Dm = kQ = kIt, (26.1)
где Dm – количество вещества, которое прореагировало, Q – количество электричества, равное произведению силы тока I на время t, k – некоторый коэффициент пропорциональности. Из уравнения (26.1) следует, что k = Dm при Q = 1, т.е. это количество прореагировавшего вещества в результате прохождения единицы количества электричества; этот коэффициент называется электрохимическим эквивалентом. Так как за единицу количества электричества можно взять разные величины (1 А.час; 1 Кл = 1 А.с; 1F = 96500 Kл), то необходимо различать электрохимические эквиваленты, которые относятся к этим трем единицам.
Второй закон Фарадея. При прохождении одного и того же количества электричества через различные электролиты количества веществ, подвергшихся превращению у электродов, пропорциональны их химическим эквивалентам А:
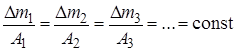 . (26.2)
. (26.2)
Если в качестве единицы количества электричества взять фарадей, то
Dm1 = k1 = A1, Dm2 = k2 = A2, Dm3 = k3 = A3, ...,
откуда в соответствии с определением k при Q = 1 F
= 1. (26.3)
Последнее уравнение дает возможность объединить оба закона Фарадея в один общий закон, согласно которому один фарадей (96500 Кл или 26,8 А.час) прошедшего электричества всегда изменяет электрохимически 1/z молей (1 моль-экв) вещества независимо от его природы.
Законы Фарадея строго выполняются для проводников второго рода. Они лежат в основе самого точного метода измерения количества электричества, прошедшего через цепь, по количеству выделенного на электроде вещества – кулонометрии (серебряный, медный, йодный, газовый кулонометры). Наблюдаемые иногда на практике отклонения от законов Фарадея являются кажущимися. Они связаны с утечками тока, протеканием неучтенных параллельных электрохимических реакций, потерями вещества при разбрызгивании растворов и т.д. В технических процессах отношение количества полученного при электролизе продукта к количеству, вычисленному по закону Фарадея, меньше единицы и называется выходом по току.
Скорость электрохимической реакции, как и скорость химической реакции, определяется как изменение количества вещества в единицу времени:
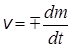 . (26.4)
. (26.4)
Но поскольку между количеством прореагировавшего вещества и количеством прошедшего электричества существует прямая пропорциональность, то на основе уравнения (26.1) можно написать, что
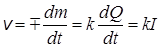 , (26.5)
, (26.5)
т.е. скорость электрохимической реакциипропорциональна силе тока. Так как для каждой реакции k является постоянной величиной, то сила тока является удобной величиной для выражения скорости любого электрохимического процесса.
Все электрохимические реакции протекают на границе раздела электрод – электролит и поэтому их скорость зависит от площади границы раздела S. В связи с этим принято относить скорость электрохимической реакции к единице поверхности раздела и определять ее как плотность тока:
i = I/S. (26.6)
На основе законов Фарадея можно рассчитать количество электричества, необходимое для получения определенного количества продукта, но не затраты электрической энергии. Затраты электрической энергии зависят от природы получаемого вещества, природы протекающей реакции и условий ее проведения. Если для получения 1 моль-экв любого вещества необходимо F кулонов электричества, то затраты электрической энергии составляют FE Вт.с. Напряжение на ванне Е для каждого процесса имеет определенное значение и может изменяться в зависимости от условий его проведения.
26.2. Поляризация электродов
Прохождение электрического тока приводит к изменениям на поверхности электродов, которые зависят от ряда факторов и в первую очередь от силы тока. Изменение электрического состояния электрода (его потенциала, плотности заряда двойного электрического слоя) под влиянием проходящего через границу раздела электрод – электролит электрического тока называется поляризациейэлектрода. При поляризации потенциал электрода отличается от равновесного значения, которое он имел бы в данных условиях при отсутствии тока.
При равновесии между электродом и раствором происходит непрерывный обмен заряженными частицами, а скорость их перехода в противоположных направлениях одинакова. Количество электричества, переходящее в этих условиях в единицу времени от электрода к раствору и обратно, называется током обмена. В случае так называемых идеально поляризуемых электродов обкладки двойного электрического слоя могут находиться в электростатическом равновесии и не обмениваться ионами в отсутствие тока. Например, ртуть, находящаяся в контакте с раствором хлорида калия, практически не отдает своих ионов раствору и двойной электрический слой не образуется. При сообщении ртутному электроду некоторого заряда от внешнего источника электрод приобретает потенциал, который может непрерывно изменяться в результате изменения сообщаемого электроду заряда, т.е. происходит поляризация электрода. До определенного значения потенциала плотность заряда на поверхности электрода непрерывно изменяется, но какой-либо электрохимический процесс отсутствует.
При прохождении тока через границу электрод – электролит остается также и ток обмена, но на него накладывается обычно значительно больший односторонний ток, который зависит от ЭДС элемента или приложенной извне разности потенциалов. При этом величина тока обмена может измениться по сравнению с равновесным значением.
Разность между потенциалом электрода под током jI и его равновесным потенциалом j называется электродной поляризацией:
Dj = jI – j. (26.7)
Потенциал jI и электродная поляризация Dj являются прежде всего функциями силы тока. В основе зависимостей jI – I и Dj – I лежат кинетические закономерности, характерные для данной электрохимической реакции. Получение зависимостей потенциал – сила (или плотность) тока (поляризационных кривых) является одним из основных методов исследования кинетики электрохимических процессов.
Любой электродный процесс представляет собой сложную гетерогенную реакцию, состоящую из нескольких последовательных стадий. На некоторых из них реакция может протекать двумя или несколькими путями. Скорость стадий и всего химического процесса зависят от состава ионопроводящей среды (природы и активности участников электродной реакции), температуры, давления, каталитических свойств границы раздела, времени от начала реакции, т.е. от тех факторов, которые определяют и скорость обычной химической реакции. Кроме того, нужно учитывать и некоторые факторы, характерные только для электрохимических процессов. Прежде всего, это потенциал электрода, который сильно влияет не только на скорость, но и направление протекания реакции и даже на природу ее продуктов. Кроме потенциала, на протекание электрохимических реакций существенное влияние оказывает и заряд электрода. Таким образом, кинетика электрохимического процесса является функцией большего числа параметров, чем кинетика обычной химической реакции. Это дает возможность более тонко и полнее регулировать скорость процесса, если известен его механизм.
Как известно из химической кинетики, скорость последовательной реакции определяется скоростью самой медленной стадии. Это положение справедливо и для электрохимических процессов. Поэтому возникновение электродной поляризации связано непосредственно с той стадией, которая определяет скорость всего процесса, т.е. с наиболее медленной стадией.
Природа и число стадий каждой электрохимической реакции зависит от ее особенностей. Как и в случае гетерогенной химической реакции, можно выделить три основных этапа – подход реагирующих частиц к границе раздела электрод – электролит, собственно электрохимический процесс, который может состоять из нескольких стадий, отвод продуктов в объем системы. Например, при электровосстановлении ионов Fe3+ до Fe2+ первой стадией является процесс доставки ионов Fe3+ к поверхности электрода путем диффузии. При контакте с электродом этот ион присоединяет электрон, превращаясь в ион Fe2+, при этом также происходит перестройка гидратной оболочки иона. Образовавшийся ион должен быть отведен от поверхности электрода, чтобы освободить место для разрядки других ионов Fe3+. Подобным же образом протекает процесс превращения ионов Sn4+ в ионы Sn2+, но в этом случае перенос электронов может происходить одновременно в одном акте или путем двух последовательных одноэлектронных переходов.
Замедленность той или стадии является непосредственной причиной поляризации электрода. Если известна природа замедленной стадии, то вместо термина поляризация можно использовать термин электродное перенапряжение, или просто перенапряжение.
 В зависимости от природы замедленной стадии можно говорить о различных видах перенапряжения. Как указывалось выше, одной из обязательных стадий любого электродного процесса является транспортировка участников реакции – их доставка (или отвод) к границе раздела электрод – электролит. Если эта стадия протекает замедленно, то концентрации участников реакции вблизи электрода изменяются по сравнению с их исходными значениями, а вместе с этим изменяется и потенциал электрода, т.е. возникает диффузионное перенапряжение (перенапряжение транспортировки). Замедленное протекание чисто химической стадии – реакции, которая предшествует акту разряда или следует за ним, также приводит к изменению концентрации участников реакции вблизи электрода, а, следовательно, и к изменению потенциала электрода. Это приводит к возникновению химического, или реакционного перенапряжения. Поляризацию, обусловленную торможением на стадиях транспортировки или химического превращения, можно назвать концентрационной поляризацией.
В зависимости от природы замедленной стадии можно говорить о различных видах перенапряжения. Как указывалось выше, одной из обязательных стадий любого электродного процесса является транспортировка участников реакции – их доставка (или отвод) к границе раздела электрод – электролит. Если эта стадия протекает замедленно, то концентрации участников реакции вблизи электрода изменяются по сравнению с их исходными значениями, а вместе с этим изменяется и потенциал электрода, т.е. возникает диффузионное перенапряжение (перенапряжение транспортировки). Замедленное протекание чисто химической стадии – реакции, которая предшествует акту разряда или следует за ним, также приводит к изменению концентрации участников реакции вблизи электрода, а, следовательно, и к изменению потенциала электрода. Это приводит к возникновению химического, или реакционного перенапряжения. Поляризацию, обусловленную торможением на стадиях транспортировки или химического превращения, можно назвать концентрационной поляризацией.
Любой электрохимический процесс включает хотя бы одну стадию, связанную с переходом электрона через границу раздела электрод – электролит. При замедленном протекании этой стадии возникает электрохимическая поляризация (замедленный разряд, перенапряжение электронного перехода). В некоторых случаях (например, при электроосаждении металлов или их растворении) замедленной стадией может быть процесс формирования или разрушения кристаллической решетки или превращение одной кристаллической модификации в другую, что приводит к фазовому перенапряжению. Концентрационные изменения вблизи поверхности электрода играют второстепенную роль при возникновении электрохимического и фазового перенапряжений. Главную роль здесь играет изменение энергии активации соответствующего процесса. Поэтому электрохимическое и фазовое перенапряжения часто объединяют под общим названием активационная поляризация.
Деление поляризации на концентрационную и активационную в определенной степени является условным. Так, фазовое перенапряжение существенно зависит от концентрации промежуточных частиц, и в этом смысле его можно отнести к концентрационной поляризации. Скорость чисто химической стадии зависит, как известно, от энергии активации, поэтому химическое перенапряжение можно рассматривать как переходное звено между концентрационной и активационной поляризациями.
В общем случае смещение потенциала электрода под током от равновесного значения является результатом наложения всех видов перенапряжения. Однако можно найти такие электродные процессы или создать такие условия, при которых преимущественное значение имеет лишь один вид перенапряжения.
26.2.1. Концентрационная поляризация
Рассмотрим в качестве примера процесс электролиза раствора нитрата серебра с концентрацией со в присутствии нитрата калия (фоновый электролит, препятствующий миграции ионов серебра к электроду под влиянием электрического поля в растворе). В качестве катода используется небольшая серебряная проволочка, анодом служит платиновый электрод с большой поверхностью. В отсутствии тока потенциал серебряного электрода описывается уравнением Нернста:
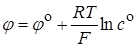 . (26.8)
. (26.8)
Приложим к электродам разность потенциалов от внешнего источника тока, достаточную для восстановления ионов серебра в металлическое серебро. Так как серебряный электрод является обратимым, то восстановление Ag+ будет происходить при очень малом отклонении потенциала серебряного электрода от равновесного значения при наложении внешней разности потенциалов. В результате восстановления ионов серебра их концентрация в непосредственной близости от поверхности катода уменьшается до некоторого значения сs, а потенциал электрода принимает новое значение
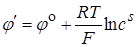 . (26.9)
. (26.9)
Так как концентрация ионов серебра в объеме раствора и вблизи анода остается постоянной, то возникает градиент концентраций, приводящий к диффузии ионов серебра из объема раствора к электродам. По мере прохождения тока градиент концентрации у катода увеличивается и диффузия ионов из раствора к электроду усиливается. Через некоторое время количество ионов, разряжающихся на катоде, становится равным количеству ионов, которые подходят к поверхности электрода в результате диффузии – устанавливается стационарное состояние.
В стационарном состоянии сила тока, проходящего через раствор, определяется количеством молей ni ионов, которые диффундируют к электроду в единицу времени. По закону Фика это количество
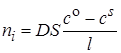 , (26.10)
, (26.10)
где D – коэффициент диффузии разряжающегося иона;
S – площадь электрода;
l – толщина диффузионного слоя, в котором происходит изменение концентрации ионов от со до сs.
Сила тока тогда равна
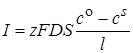 . (26.11)
. (26.11)
При увеличении силы тока число разряжающихся в единицу времени ионов возрастает, а их концентрация вблизи поверхности электрода уменьшается. Если скорость разряда ионов становится больше скорости диффузии, то концентрация сs ® 0, а сила тока достигает некоторого предельного значения, которое называется предельным током диффузииIд:
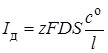 . (26.12)
. (26.12)
Из двух последних уравнений следует, что
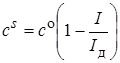 . (26.13)
. (26.13)
Подставив это значение в уравнение (26.9) и вычтя из него уравнение (26.8), что смещение потенциала, обусловленное концентрационной поляризацией
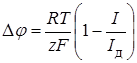 . (26.14)
. (26.14)
Величины Dj обоих электродов представляют электродвижущую силу концентрационной поляризации, которая направлена против приложенной разности потенциалов от внешнего источника. Возникновение ЭДС концентрационной поляризации приводит к увеличению расхода электрической энергии в различных процессах промышленного электролиза, снижает ЭДС химических источников тока при их работе. Одним из основных методов снижения концентрационной поляризации является перемешивание раствора в электролитической ванне или создание особых условий эксплуатации химических источников тока при их работе.
26.2.2. Электрохимическая поляризация
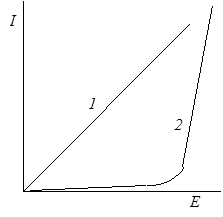 Рис. 26.1 Зависимость силы тока от напряжения для проводников первого (1) и второго рода (2)
Рис. 26.1 Зависимость силы тока от напряжения для проводников первого (1) и второго рода (2)
|
Протекание электрического тока через электролиты, а также разложение веществ (электролиз) происходит только при достижении определенной величины напряжения. При постепенном увеличении напряжения от внешнего источника сила тока в цепи остается незначительной до тех пор, пока напряжение не достигнет определенного значения. После этого происходит быстрое нарастание силы тока и наступает собственно электролиз (рис. 26.1, кривая 2). Это противоречит закону Ома в обычной форме, согласно которому сила тока в цепи всегда пропорциональна напряжению: I = E/R (рис. 26.1, прямая 1).
Для объяснения такого характера зависимости силы тока от напряжения рассмотрим следующий пример. Два платиновых электрода опущены в электролитическую ванну с раствором соляной кислоты. При отсутствии напряжения от внешнего источника потенциалы обоих электродов равны. При подаче даже небольшого напряжения во внешней цепи между электродами потечет электрический ток – к одному из электродов будут поступать электроны, а из другого уходить. Электроны не могут непосредственно проходить через раствор электролита, поэтому при отсутствии электрохимических процессов на одном из электродов происходит увеличение числа электронов (электрод заряжается отрицательно), на другом – уменьшение (электрод заряжается положительно). Возникает разность потенциалов между электродами, которая направлена против напряжения от внешнего источника. Когда эта разность потенциалов становится равной приложенной извне разности потенциалов, ток в цепи прекратится. При увеличении внешнего напряжения происходит дальнейший процесс заряжения и изменения потенциалов электродов до тех пор, пока поляризация не приведет к протеканию электрохимических процессов, сопровождающихся выделением и поглощением электронов. Начинается собственно электролиз и через систему протекает стационарный ток. В этом случае полностью проявляется электрохимическая (химическая) поляризация, ЭДС которой направлена против внешней разности потенциалов.
В рассматриваемом примере на отрицательно заряженном электроде (катоде) электрохимическим процессом является процесс разряда ионов гидроксония:
2H3O+ + 2e ® 2H2O + H2
Эта реакция характерна для водородного электрода. Равновесию между ионами гидроксония и газообразным водородом соответствует вполне определенный потенциал, величина которого зависит от активности ионов гидроксония, температуры, давления водорода. Очевидно, что разряд ионов возможен лишь при достижении этого равновесного потенциала водородного электрода (при отсутствии перенапряжения). Этим и определяется предельное значение поляризации катода при электролизе.
На аноде при достижении определенного потенциала происходит выделение хлора. Этому процессу также соответствует потенциал хлорного электрода, зависящий от активности хлорид-ионов, температуры, давления хлора.
Таким образом, при электролизе возникает электрохимическая цепь
Pt(H2)| HCl |(Cl2)Pt,
ЭДС которой направлена против внешней разности потенциалов. Это и приводит к кажущемуся противоречию с законом Ома, который в этом случае должен быть записан в форме
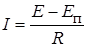 , (26.15)
, (26.15)
где Еп – ЭДС поляризации.
Вычислить величину Еп можно по уравнению Нернста (в отсутствие концентрационной поляризации):
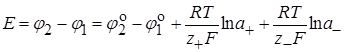 . (26.16)
. (26.16)
Таким образом, разложение веществ путем электролиза может происходить лишь при определенном напряжении, зависящем от природы электролита и условий проведения электролиза.
26.2.3. Напряжение разложения
Минимальная разность потенциалов, которую нужно создать между электродами, чтобы начался электролиз, называется напряжением разложенияэлектролита. Эта величина равна сумме потенциалов разряда ионов на электродах. В отсутствии перенапряжения на электродах напряжение разложения равно сумме равновесных потенциалов электродов, образующихся после начала электролиза. В приведенном выше примере электролиза водного раствора соляной кислоты оно равно сумме равновесных потенциалов водородного и хлорного электродов.
Величина напряжения разложения более или менее точно определяется для электролита при заданных условиях лишь в случае выделения на электродах индивидуальных твердых веществ, например, чистых металлов. Если в результате электролиза образуются твердые или жидкие растворы и, особенно, газы, напряжение разложения зависит от формы и размеров электродов, характера их поверхности, условий отвода газов и др. Таким образом, напряжение разложения данного электролита не является для него однозначной характеристикой и зависит от условий проведения электролиза. ЭДС электрохимической поляризации представляет собой ту реальную ЭДС, которая возникает при наложении внешней разности потенциалов и препятствует электролизу. В некоторых случаях предельная поляризация электродов может быть незначительно меньшей, чем приложенная разность потенциалов. Тогда эта разность и является величиной напряжения разложения. Следовательно, минимальная величина напряжения разложения равна ЭДС поляризации.
В таблице 26.1 в качестве примера приведены предельные ЭДС поляризации водных растворов некоторых электролитов средних концентраций при электролизе на платиновых электродах.
Из таблицы видно, что предельная ЭДС поляризации при электролизе кислородсодержащих кислот и щелочей примерно одинаковы. Это может быть объяснено тем, что в разных растворах при электролизе протекают одни и те же процессы. Действительно, при электролизе этих растворов на катоде происходит выделение водорода, а на аноде – кислорода. В растворах кислот выделение водорода происходит по схеме
2Н3О+ + 2е ® 2Н2О + Н2,
т.е. разряжаются ионы гидроксония.
В растворах щелочей также возможен их разряд, но в таких растворах их концентрация очень мала. При большой силе тока, который переносится главным образом ионами щелочных металлов, к электроду может подойти лишь незначительное количество ионов гидроксония, которое не может обеспечить выделение заметных количеств водорода. Видимо, выделение водорода происходит за счет разложения молекул воды, адсорбированных на электроде:
2H2O + 2e ® 2 OH– + H2
Таблица 26.1
Предельная ЭДС поляризации при электролизе растворов некоторых веществ
| Электролит | Предельная ЭДС поляризации, В | Электролит | Предельная ЭДС поляризации, В |
| H2SO4 | 1,67 | AgNO3 | 0,70 |
| HNO3 | 1,69 | HCl 2 М | 1,26 |
| H3PO4 | 1,70 | HCl 1 М | 1,31 |
| NaOH | 1,69 | HCl 0,5 М | 1,34 |
| KOH | 1,67 | HCl 1/6 М | 1,41 |
| NH4OH | 1,74 | HCl 1/16 М | 1,62 |
| Na2SO4 | 2,21 | HCl 1/32 М | 1,69 |
| ZnSO4 | 2,35 |
Кислород при электролизе растворов щелочей выделяется в результате разряда ионов гидроксила на аноде:
4ОН– – 4е ® 2Н2О + О2
При электролизе растворов кислот, где концентрация ионов гидроксила очень мала, кислород выделяется в результате разложения молекул воды:
6Н2О – 4е ® 4Н3О+ + О2
При больших потенциалах (2,5 – 3 В) возможны и другие анодные процессы. Например, при электролизе растворов хлорной кислоты начинается выделение кислорода из молекул HClO4.
Таким образом, при электролизе растворов кислот, щелочей, а также соответствующих солей щелочных и щелочноземельных металлов на электродах протекает первичный процесс разложения воды. Роль остальных ионов сводится к обеспечению достаточной электропроводности растворов.
Для растворов других веществ ЭДС поляризации отличаются, что указывает на различный характер электродных процессов для разных веществ. В растворах солей металлов, менее электроотрицательных, чем водород, на катоде может выделяться металл. При электролизе растворов кислот, молекулы которых не содержат кислород, на аноде обычно разряжаются анионы. Интересно в этом отношении поведение соляной кислоты: в концентрированных растворах на аноде выделяется хлор, а в разбавленных – кислород, при этом изменяется ЭДС поляризации. Это связано с тем, что при разбавлении кислоты уменьшается активность хлорид-ионов. Равновесный потенциал хлорного электрода становится более положительным, чем потенциал разряда ионов ОН–, поэтому изменяется анодный процесс – уменьшается разряд ионов хлора и происходит разряд ионов гидроксила или молекул воды.
26.3. Перенапряжение
Во многих случаях для того, чтобы начался электролиз, к электролитической ванне необходимо приложить напряжение, превосходящее на некоторую конечную величину ЭДС электрохимической поляризации. Разность между напряжением разложения и суммой равновесных потенциалов электродов называется перенапряжением. Таким образом, перенапряжения h на катоде и аноде равны:
hА = j – j А, (26.17)
hК = j ¢– j К, (26.18)
где j и j¢ – потенциалы выделения на аноде и катоде, jА и jК – соответствующие равновесные потенциалы.
Величина перенапряжения на электроде зависит от природы электрода, состава раствора, плотности тока и других факторов. Перенапряжение в целом в электролитической ванне равно тому избыточному напряжению, которое нужно приложить к ванне сверх ее равновесной ЭДС, чтобы начался электролиз. Избыточное напряжение включает, кроме перенапряжения на электродах, также падение напряжения на внутреннем сопротивлении, которое зависит от электрического сопротивления раствора.
В 1905 г. Тафельполучил эмпирическое уравнение зависимости перенапряжения от плотности тока, т.е. от скорости электрохимического процесса:
h = а + blg i, (26.19)
где h – перенапряжение, В; i – плотность тока, а/см2; a и b – константы.
Величина а равна перенапряжению при силе тока 1 а/см2. Она существенно зависит как от природы электрода, так и раствора. Величина b мало зависит от природы электрода и определяется характером самого электрохимического процесса. Она равна приблизительно 2.2,3 RT/zF, т.е. около 0,116/z В при комнатной температуре.
В таблице 26.2 приведены величины перенапряжения для процесса катодного выделения водорода на некоторых металлических электродах.
Реакция выделения водорода имеет большое практическое значение во многих электрохимических процессах – электролиз воды и различных водных растворов, хлорный электролиз, работа химических источников тока, процессы коррозии и др., поэтому перенапряжение водорода изучено наиболее подробно.
При выделении водорода из растворов минеральных кислот и оснований, а также из водных растворов солей при малых отклонениях от равновесного потенциала наблюдается линейная зависимость между перенапряжением и плотностью тока. С увеличением плотности тока зависимость выражается уравнением Тафеля и для некоторых металлов постоянные а и b сохраняют свое значение до высоких плотностей тока.
Таблица 26.2.
Значения констант а и b уравнения Тафеля для реакции
катодного выделения водорода на разных металлах при 293 К
| Металл | Раствор | а | b |
| Свинец | H2SO4 0,5 M | 1,533 | 0,118 |
| Таллий | H2SO4 0,85 M | 1,55 | 0,140 |
| Ртуть | H2SO4 2,5 M | 1,400 | 0,116 |
| Ртуть | HCl 1,0 M | 1,390 | 0,119 |
| Ртуть | KOH 0,1 M | 1,430 | 0,093 |
| Кадмий | H2SO4 0,85 M | 1,450 | 0,120 |
| Цинк | H2SO4 0,5 M | 1,24 | 0,118 |
| Олово | HCl 1,0 M | 1,24 | 0,116 |
| Медь | HCl 0,1 M | 0,790 | 0,117 |
| Серебро | HCl 1,0 M | 0,320 | 0,060 |
| Серебро | HCl 5,0 M | 0,470 | 0,070 |
| Железо | HCl 1,0 M | 0,770 | 0,130 |
| Железо | NaOH 4,8 M | 0,350 | 0,070 |
| Никель | NaOH 0,11 M | 0,64 | 0,100 |
| Кобальт | HCl 1,0 M | 0,62 | 0,140 |
| Палладий | KOH 0,1 M | 0,637 | 0,125 |
| Вольфрам | HCl 1,0 M | 0,23 | 0,040 |
| Вольфрам | HCl 5,0 M | 0,550 | 0,110 |
| Платина | HCl 0,5 M | 0,073 | 0,028 |
Для других металлов (свинец, платина) в определенной области плотностей тока наблюдается переход к новой полулогарифмической зависимости. При исследовании перенапряжения водорода на ртутных катодах из растворов чистых кислот установлено, что перенапряжение зависит от рН лишь при концентрациях кислоты больше 0,1 моль/л, а в присутствии посторонних электролитов – при меньших концентрациях кислот. В щелочной области в растворах органических оснований типа NR4OH, где процесс не осложнен выделением щелочного металла, перенапряжение водорода уменьшается при увеличении концентрации основания. Таким образом, максимальное перенапряжение наблюдается в нейтральных растворах.
Перенапряжение водорода снижается с повышением температуры, при этом температурный коэффициент зависит от природы металла и плотности тока.
26.3.1. Теории водородного перенапряжения
Перенапряжение является следствием относительно малой скорости протекания электрохимического процесса. Как отмечалось выше, электрохимические реакции являются сложными процессами, состоящими из нескольких последовательных стадий, и малая скорость любой из них приводит к перенапряжению.
Разряд ионов гидроксония можно представить следующими стадиями:
1. Диффузионная стадия. Так как процесс разряда происходит на поверхности электрода, необходима быстрая доставка ионов Н3О+ к этой поверхности, что и осуществляется за счет переноса ионов и их диффузии.
2. Стадия дегидратации. Разряд иона гидроксония невозможен без предварительной дегидратации:
H3O+ ® H+ + H2O.
3. Стадия разряда и адсорбции протона. Собственно электрохимический процесс заключается в присоединении электрона Н+ + е ® Н, а образовавшиеся атомы водорода при этом адсорбируются на поверхности металла:
Н + Ме ® Надс(Ме)
4. Стадия рекомбинации (молизации). При насыщении поверхности электрода адсорбированные атомы водорода молизуются и переходят в раствор:
2Н(адс) ® Н2(раствор)
5. Стадия выделения газа. Образующиеся молекулы водорода уходят с поверхности электрода в объем в виде пузырьков газа:
Н2(раствор) ® Н2(газ)
Было предложено ряд теорий водородного перенапряжения, которые отличаются друг от друга тем, какая из стадий разряда считается наиболее медленной. Так, по Мюллеру лимитирующей считается последняя стадия, по Леблану – стадия дегидратации, по Смитсу – стадия разряда иона, по Тафелю – процесса молизации, по Нернсту – стадия адсорбции. Справедливость той или иной теории определяется ее возможностью объяснить уравнение Тафеля, которое получено на основании экспериментальных данных. Однако все основные теории при определенных предположениях приводят к формуле Тафеля.
Рекомбинационная теорияпредложена Тафелем еще в 1905 г. Согласно этой теории, наиболее медленной является стадия молизации адсорбированного водорода, поэтому в процессе электролиза концентрация атомарного водорода на поверхности электрода увеличивается по сравнению с равновесной с молекулярным водородом (газ), что и приводит к сдвигу потенциала электрода в отрицательную сторону. В соответствии с уравнением
Н(адс) + Н2О ® Н3О+ + е
увеличение концентрации адсорбированного водорода сдвигает электродное равновесие вправо. Скорость ионизации водорода при этом увеличивается, а так как электроны остаются на электроде, то его отрицательный потенциал увеличивается.
Скорость v рекомбинации атомов водорода в молекулы (2H ® H2) пропорциональна квадрату поверхностной концентрации [H] адсорбированного на электроде водорода:
v = k¢[Н]2. (26.20)
С другой стороны, скорость электрохимического выделения водорода v¢ определяется силой тока I:
v¢ = I/2F. (26.21)
Если лимитирующей стадией является стадия молизации, т.е. скорость vзначительно меньше скоростей всех других стадий, то и v¢ = v, из чего следует, что
I/F = 2k¢[Н]2 = k[Н]2. (26.22)
Так как перенапряжение обусловлено увеличением концентрации адсорбированного водорода, то по Тафелю
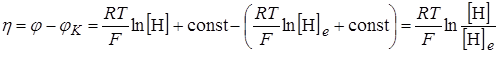 , (26.23)
, (26.23)
где [H]e – поверхностная концентрация атомарного водорода на электроде при условии равновесия этого водорода с молекулярным водородом в газовой фазе.
Если подставить в последнее уравнение значение [H], выраженное через силу тока, то для перенапряжения получим:
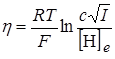 , (26.24)
, (26.24)
где с = 1/(2k¢F)1/2
Так как с и [H]e являются постоянными величинами, то уравнение можно записать в виде
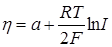 (26.25)
(26.25)
или при 298 К
h = а + 0,029 lgI. (26.26)
Это уравнение по форме совпадает с уравнением Тафеля (26.17), но коэффициент b оказывается в четыре раза меньше опытной величины 0,116.
В дальнейшем эта теория была развита рядом ученых (Гориучи, Темкин, Кобозев, Антропов и др.). В этих работах было учтено влияние неоднородности поверхности и сил взаимодействия между адсорбированными атомами на величину перенапряжения. В отличие от первоначальной теории, согласно которой коэффициент b постоянен для всех металлов, в действительности он оказывается функцией природы металла и может иметь различные значения. Однако эти уточнения не изменяют саму форму уравнения Тафеля.
Теория медленного разряда ионов. Согласно этой теории наиболее медленной стадией электрохимического выделения водорода является процесс разряда ионов. Хотя эта идея высказывалась давно, теория привлекла внимание лишь после работ Эрдей-Груса и Фольмера (1930 г.), которые предположили, что разряд ионов требует значительной энергии активации и поэтому может происходить с малой скоростью. На основании этого предположения можно вывести уравнение Тафеля.
Рассмотрим скорость электрохимического процесса как скорость обычной химической реакции, которая зависит от концентрации реагирующих частиц:
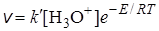 . (26.27)
. (26.27)
Скорость электрохимического процесса определяется силой тока:
It = zFn; Idt =zFdn; v = dn/dt = I/zF, (26.28)
тогда сила тока
I = 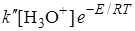 , (26.29)
, (26.29)
или для раствора постоянного состава
I = 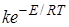 . (26.30)
. (26.30)
Роль перенапряжения сводится к снижению энергии активации. Если положить, что это снижение пропорционально перенапряжению и равно aFh (a – коэффициент пропорциональности), то энергия активации
Е = Eo – aFn, (26.31)
где Ео – энергия активации неполяризованного электрода.
Тогда, при наличии перенапряжения, сила тока
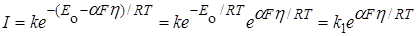 . (26.32)
. (26.32)
Логарифмируя это уравнение, получим:
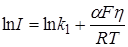 . (26.33)
. (26.33)
Отсюда величина перенапряжения
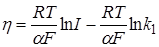 . (26.34)
. (26.34)
Силу тока выразим через его плотность: I = is. Считая поверхность электрода s постоянной, переходя к десятичным логарифмам и обозначая сумму постоянных при данной температуре слагаемых через а, получаем уравнение Тафеля:
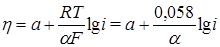 , (26.35)
, (26.35)
где b = 0,058/a. Если a = 0,5, то b = 0,116.
Теория замедленного разряда, как и другие современные теории, объясняет, почему формула Тафеля неприменима при малых поляризациях электрода (при малых плотностях тока). Уравнение (26.30) справедливо при условии, что процесс идет в одном направлении, т.е. происходит только электровосстановление или электроокисление. Но при электролизе сохраняется ток обмена, обратный пропускаемому через ячейку току, поэтому необходимо учитывать и обратный электролизу процесс. Тогда сила тока
I = 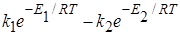 , (26.36)
, (26.36)
где индексы 1 и 2 относятся к прямому и обратному процессам.
При поляризации электрода, т.е. при повышении его отрицательного потенциала, скорость прямого процесса увеличивается, а обратного уменьшается, и при некотором перенапряжении последняя становится настолько малой, что ею можно пренебречь, и тогда выполняется уравнение Тафеля. Анализ уравнения (26.35) показывает также, что вблизи равновесного потенциала (при малых перенапряжениях) зависимость I – h должна быть линейной.
Теория Фольмерав первоначальном виде не учитывала строения двойного электрического слоя на границе электрод – раствор, поэтому не могла объяснить влияния состава электролита на величину перенапряжения водорода. В дальнейшем в работах А. Н. Фрумкина было показано, что силы электростатического взаимодействия между электродом и ионами вызывают изменение концентрации реагирующих ионов в приэлектродном слое и что наличие двойного электрического слоя влияет на величину энергии активации электродного процесса.
В настоящее время считается, что в зависимости от природы металла, условий проведения электролиза лимитирующей может быть как стадия рекомбинации, так и стадия разряда ионов, поэтому не имеет смысла противопоставлять разные теории перенапряжения.
26.3.2. Кинетика электролитического выделения кислорода
Как теоретически, так и практически большой интерес представляет процесс электрохимического выделения кислорода. Вследствие высокой химической активности кислорода процесс осложняется образованием на поверхности электрода различных оксидов, поэтому выделение газообразного кислорода происходит обычно с окисленной поверхности. В связи со сложностью процесса до сих пор нет достаточно обоснованной теории кислородного перенапряжения.
В зависимости от состава раствора процесс электрохимического выделения кислорода может происходить по-разному. При электролизе растворов щелочей наиболее вероятным источником анодного кислорода являются ионы гидроксила. Суммарное уравнение реакции разряда ионов можно представить как
4ОН- ® О2 + Н2О +4е
В кислых растворах, где содержание ионов ОН- мало и не может обеспечить необходимую скорость образования кислорода, разряжению подвергаются молекулы воды:
2H2O ® O2 + 4H+ +4e
Из нейтральных солевых растворов кислород может выделяться в результате разряжения гидроксильных ионов и молекул воды. Преобладать будет тот процесс, который в данных условиях требует меньшей энергии. В концентрированных растворах кислородсодержащих кислот при высоких плотностях тока в реакциях выделения кислорода могут принимать непосредственное участие анионы кислот. Например, при электролизе раствора серной кислоты кислород выделяется в результате разряжения сульфат-ионов в реакции
2 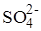 ® 2SO3 + O2 + 4e
® 2SO3 + O2 + 4e
с последующей регенерацией ионов :
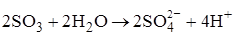
Кислород выделяется всегда при потенциалах более положительных, чем потенциал обратимого кислородного электрода в данных условиях. Эта разница между потенциалом электрода под током и его равновесным потенциалом и представляет собой перенапряжение кислорода. Выяснение кинетического механизма анодного выделения кислорода представляет собой сложную задачу, решение которой связано не только со значительными экспериментальными трудностями, но и с большим количеством теоретически возможных вариантов протекания процесса.
В реакции электролитического образования кислорода независимо от того, в какой среде она происходит, принимают участие не два, как в реакции выделения водорода, а четыре электрона. Этим обусловлено появление нескольких электрохимических стадий, каждая из которых может определять скорость всего электрохимического процесса. Наряду с этим нужно учитывать возможность замедленного протекания рекомбинации и электрохимической десорбции. Наконец, поскольку выделение кислорода происходит обычно на поверхности металла, степень окисленности которого зависит от потенциала и времени электролиза, образование и распад оксидов также могут влиять на кинетику процесса. Предложены различные кинетические схемы анодного выделения кислорода. Например, при выделении кислорода из щелочных растворов возможны следующие варианты (в приведенных схемах OH· – гидроксильный радикал):
I вариант IІ вариант
1. 2OH- ® 2OH· + 2e 1. 2OH- ® 2OH· + 2e
2. 2OH· + 2OH- ® 2O- + 2H2O 2. 2OH· + 2OH- ®2O- + 2H2O
3. 2O- ® 2O + 2e 3. 2O- + 2MOx ® 2MOx+1 +2e
4. 2O ® O2 4. 2MOx+1 ® 2MOx + O2
III вариант IV вариант
1. 4OH- + M ® 4MOH + 4e 1. 2OH- ® 2OH· + 2e
2. 4MOH ® 2MO + 2M + 2H2O 2. 2OH· + 2OH- ® 2H2 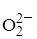
3. 2MO ® 2M + O2 3. 2H2 ® + 2H2O
4. ® O2 +2e
Приведенные схемы не исчерпывают всех возможных вариантов протекания реакции образования кислорода. Можно предположить, что уже на первой стадии разряд гидроксил-ионов приводит к образованию атомарного кислорода или неустойчивого поверхностного оксида:
2OH- ® O + H2O + 2e
или
2OH- + MOx ® MOx+1 + H2O + 2e
Любая из приведенных стадий может быть замедленной и определять скорость всей реакции. Для того чтобы сделать правильный выбор между теоретически возможными случаями и установить истинные причины кислородного перенапряжения, необходимо использовать критерии общей теории кинетики электродных процессов. Одним из таких критериев может быть, например, величина наклона зависимостей h – lgi. Однако и в этом случае заключения имеют вероятностный характер, поскольку величина наклона b не может считаться достаточным критерием окончательного выбора механизма. Кроме того, в таком сложном процессе, каким является электрохимическое выделение кислорода, почти всегда есть возможность параллельного протекания нескольких стадий с близкими скоростями.
26.7. Полярография
Наиболее распространенным методом исследования электрохимических процессов является установление зависимости между силой тока и напряжением – вольтамперометрия. Первым по времени появления и одним из наиболее распространенных методов вольтамперометрии является полярография. Полярографический метод разработан чешским ученым Я.Гейровским (1922). Он заключается в проведении электролиза исследуемого раствора в электролитической ячейке и снятии кривой зависимости силы тока от подаваемого на ячейку напряжения – полярограммы.
Простейшая схема полярографической установки (полярографа) представлена на рисунке 26.2. В сосуде В находится исследуемый раствор, в который погружен стеклянный капилляр D, через который под давлением ртутного столба медленно вытекает ртуть из сосуда С, высоту подъема которого можно регулировать. Образующиеся на конце капилляра ртутные капли через равные промежутки времени (период капания, он составляет обычно несколько секунд) отрываются от капилляра и падают на дно сосуда.
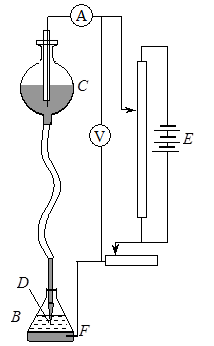 Рис. 26.2. Схема полярографической установки
Рис. 26.2. Схема полярографической установки
|
От внешнего источника тока Е (аккумулятора) на ячейку подается определенное напряжение, величину которого можно регулировать с помощью реостата и измерять вольтметром V. Каждая ртутная капля до момента ее отрыва служит электродом. Капельный ртутный электрод может служить катодом при изучении процессов электровосстановления или анодом при электроокислении. В качестве вспомогательного электрода F обычно применяют ртутный электрод с большой поверхностью, потенциал которого практически не изменяется при прохождении тока небольшой плотности (на рисунке это донная ртуть). Силу проходящего через ячейку тока измеряют с помощью чувствительного амперметра А.
При прохождении тока в общем случае изменяются потенциалы обоих электродов, а также часть приложенного напряжения падает из-за сопротивления раствора:
Е = j А – j K + IR, (26.37)
где Е – внешняя разность потенциалов, jA и jК – потенциалы анода и катода, I – сила тока, R – омическое сопротивление раствора.
Сила тока в ячейке обычно невелика (10-5 – 10-6 А), поэтому практически все изменение напряжения при переходе от одного значения тока к другому обусловлено изменением потенциала капельного микроэлектрода, поверхность которого на несколько порядков меньше поверхности донной ртути. К исследуемому раствору добавляют также избыток индифферентного электролита (фоновый электролит) для обеспечения высокой электрической проводимости раствора, поэтому падение напряжения в ячейке можно не учитывать. Таким образом, можно считать, что
Е =j K + const, (26.38)
т.е. кривая зависимости сила тока напряжение фактически является кривой зависимости силы тока от потенциала капельного электрода. Если в растворе находятся частицы, способные восстанавливаться на ртутном электроде, то, снимая зависимость I – E, получают полярографическую кривую, или полярограмму (рис. 26.3).
Рассмотрим явление концентрационной поляризации на капельном ртутном электроде при разряде ионов металла. При таком разряде на ртутном электроде происходит образование амальгамы металла, и потенциал электрода
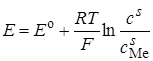 , (26.39)
, (26.39)
где cs – концентрация амальгамы металла вблизи поверхности электрода; 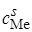 – концентрация ионов металла в растворе около электрода.
– концентрация ионов металла в растворе около электрода.
Чтобы найти уравнение концентрационной поляризации на капельном ртутном электроде, необходимо определить величины cs и в зависимости от протекающего тока.
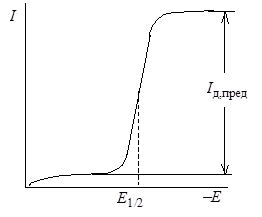 Рис. 26.3. Полярографическая кривая
Рис. 26.3. Полярографическая кривая
|
Процесс диффузии к растущей сферической поверхности значительно сложнее процесса диффузии к неподвижному твердому электроду. Так как поверхность ртутной капли в процессе ее формирования возрастает от очень малой величины (в момент, непосредственно следующий за отрывом предыдущей капли) до некоторого максимального значения (в момент, предшествующий отрыву данной капли), то и диффузионный ток периодически меняется. Следовательно, мгновенный ток отличается от его среднего значения за период образования капли. Среднее значение тока (А.см-2) описывается уравнением Ильковича:
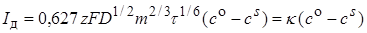 , (26.40)
, (26.40)
где D – коэффициент диффузии;
m – масса ртути, вытекающий в единицу времени (мг.с-1);
t – период капания (с);
со и сs – концентрация ионов металла в объеме раствора и вблизи поверхности электрода (моль/см3);
k – коэффициент, постоянный при заданном режиме работе полярографической установки.
Когда скорость разряда ионов на электроде становится больше скорости подхода ионов к поверхности электрода за счет диффузии, концентрация сs становится равной нулю, а диффузионный ток достигает величины предельного диффузионного тока Iд,пред:
Iд,пред = kсо (26.26)
По уравнению Ильковича можно определить число электронов z, принимающих участие в электродной реакции, коэффициенты диффузии и концентрации веществ. Уравнения (26.40 – 26.41) могут не только являться основой количественного анализа, но и служить для прямого расчета концентрации определяемого вещества по предельному току, если известен коэффициент k. Однако расчетные значения k не являются достаточно надежными, поэтому на практике при количественном анализе проводят калибровку полярографической установки по стандартным растворам.
Качественный полярографический анализ основан на уравнении Гейровского – Ильковича:
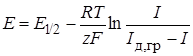 , (26.42)
, (26.42)
где Е1/2 – потенциал полуволны, соответствующий случаю, когда I = Iд,пред/2. Потенциал полуволны определяется только природой восстанавливаемого вещества и не зависит от его концентрации.
Кроме капающего ртутного электрода в полярографии используется также вращающийся дисковый электрод (платиновый). Его особенность заключается в том, что в любой точке его поверхности диффузионный слой имеет постоянную толщину. Большие возможности для исследований дает комбинированный электрод из диска и кольца. Диск и кольцо закрепляются в твердом изоляционном материале и образуют вместе один вращающийся электрод с двумя независимыми токоподводами. Это дает возможность осуществлять раздельную поляризацию диска и кольца. Поэтому вещество, которое окисляется на диске, может восстанавливаться на кольце и наоборот. Благодаря этому можно устанавливать природу продуктов электродной реакции, их константы устойчивости и т.д.
Полярографический метод позволяет достаточно быстро проводить анализ сложных систем с малым содержанием веществ (до 10-5 моль/л). Он применяется для определения многих органических соединений, растворенных газов (например, кислорода). Кроме того, полярография является одним из методов исследования кинетики и механизма электродных процессов.
26.5. Электрохимическое выделение металлов
Электрохимическое выделение металлов из водных растворов их соединений лежит в основе гидроэлектрометаллургических процессов – получения металлов из руд (электроэкстракция) и их очистки (рафинирование) при электролизе. Электрохимическое выделение металлов используется для защиты основного металла от разрушения (коррозии) путем нанесения покрытий из других металлов или сплавов. Этот процесс используется также для придания изделиям декоративного вида (гальванотехника), получения копий художественных предметов, изготовления печатных плат и т.п. (гальванопластика). Возможность использования электролиза в практических целях была открыта академиком Б.С.Якоби еще в 1837 г.
Электролитическое выделение металлов чаще всего осуществляется из растворов их простых солей – сульфатов, хлоридов, нитратов. Суммарной катодной реакцией является при этом разряжение гидратированных ионов металлов и их последующим переходом в кристаллическую решетку образующегося на катоде осадка:
Mz+. xH2O + ze ® Mтв + xH2O
Поляризация при электрохимическом выделении металлов, как и при других электродных процессах, зависит от плотности тока, но характер этой зависимости может быть различным в разных диапазонах плотности тока, а также меняться во времени. Характер осадка и условия его формирования во времени зависят не только от природы металла, но и от состава раствора и присутствия в нем различных примесей (например, поверхностно-активных веществ и окислителей, в том числе растворенного кислорода), которые влияют на кинетику электровыделения металла.
При электролизе растворов простых солей характер катодных осадков и величина катодной поляризации определяются, прежде всего, природой выделяемого металла. Можно выделить три группы металлов по величине перенапряжения и качеству образуемых осадков.
К первой группе относятся металлы, которые выделяются из водных растворов без перенапряжения (ртуть) или с очень малым перенапряжением (несколько милливольт) – серебро, таллий, свинец, кадмий, олово. При промышленных плотностях тока эти металлы образуют довольно грубые осадки – средний размер зерна равен 10-3 см и больше. Токи обмена для металлов этой группы очень большие – 102 – 103 А.м-2.
Для второй группы металлов (висмут, медь, цинк) характерно перенапряжение порядка нескольких десятков милливольт, образование более тонких осадков (средний размер зерна 10-4 – 10-3 см), меньшие токи обмена – порядка 10-1 А.м-2.
Металлы третьей группы (кобальт, железо, никель) выделяются с наибольшим перенапряжением – несколько десятых вольта. Они образуют плотные тонкокристаллические осадки с размером зерен порядка 10-5 см и меньше. Токи обмена для них очень малы – порядка 10-4 – 10-5 А.м-2.
При катодном восстановлении металлов из растворов их простых солей существенное значение имеет природа аниона. Особенно сильно влияние аниона проявляется для металлов, выделение которых не сопровождается высокой поляризацией. Обычно перенапряжение снижается в таком порядке:

причем в этом же направлении возрастает тенденция к образованию более грубых крупнокристаллических осадков.
Присутствие в растворе “индифферентных” катионов обычно увеличивает перенапряжение металлов. Такие эффекты наблюдаются при выделении никеля, цинка, меди и других металлов. В водных растворах обычными “посторонними” ионами являются ионы водорода. Увеличение их концентрации приводит чаще всего к повышению перенапряжения. Значительное его возрастание наблюдается также в присутствии поверхностно-активных катионов типа тетразамещенных ионов аммония.
Высокая чувствительность процесса электроосаждения металлов к чистоте растворов свидетельствует о том, что присутствие не только электролитов, но и каких-либо других веществ, особенно тех, которые обладают поверхностной активностью, должно играть здесь существенную роль. Введение в раствор небольших количеств молекулярных и ионных веществ является одним из наиболее эффективных способов влияния на ход процесса электровосстановления металлов. Многие, главным образом органические, вещества способны усиливать блеск осадков, сглаживать их поверхность, изменять пористость, твердость и т.д.
Электродная поляризация, которая наблюдается при выделении металлов, связана или с фазовыми превращениями и может быть одним из видов фазового превращения (замедленность образования трехмерных и двумерных зародышей, поверхностная диффузия), или с замедленностью собственно электрохимической стадии. При осаждении металлов существенную роль играют трудности на стадии транспортирования, а также на стадии химического превращения, которое предшествует электрохимическому акту. Поэтому при рассмотрении процессов катодного выделения металлов нужно всегда учитывать концентрационную поляризацию, т.е. диффузионное перенапряжение и химическое или реакционное перенапряжение. Наконец, в условиях катодного выделения металлов энергетическое состояние иона в образующемся осадке может отличаться от его состояния в нормаль
| <== предыдущая лекция | | | следующая лекция ==> |
| Уравнение плоской волны, движущейся в произвольном направлении. Фазовая скорость и скорость распространения энергии | | | Поле электрического диполя. |
Дата добавления: 2015-12-16; просмотров: 4949;