Вакцины 243
зуемых при создании вакцин, состоит в размещении протективного антигена патогенной бактерии на поверхности живой непатогенной бактерии. Многие бактерии имеют жгутики, состоящие из белка флагеллина; под микроскопом они выглядят как нити, отходящие от бактериальной клетки. Если сделать так, что жгутики непатогенного микроорганизма будут нести специфический эпитоп патогенного микроорганизма, то можно будет индуцировать выработку протективных антител.
Именно такой подход использовали при создании противохолерной вакцины. Синтетический олигонуклеотид, кодирующий эпитоп субъединицы В холерного токсина, встроили в гипервариабельный участок гена флагеллина Salmonella и полученную конструкцию ввели в дефектный по флагеллину штамм Salmonella. При этом было известно, что эпитоп, включающий 50—64 аминокислотные остатки субъединицы В холерного токсина, индуцирует выработку антител к интактному холерному токсину. Химерный флагеллин нормально функционировал, а эпитоп холерного токсина размещался на поверхности жгутиков. Иммунизация мышей с помощью интраперитонеальной инъекции примерно 5-106 живых или убитых формалином бактерий с модифицированным флагеллином индуцировала выработку большого количества антител как к пептиду (50—64 аминокислотным остаткам), так и к молекуле интактного холерного токсина. Аналогичным образом можно встраивать два и даже три разных эпитопа в один флагеллиновый ген Salmonella и создать поливалентную противобактериальную вакцину.
С помощью перорального введения аттенуированных штаммов Salmonella можно осуществлять доставку в организм хозяина многих бактериальных, вирусных и паразитарных антигенов. Большую роль при этом играет выбор промотора, контролирующего транскрипцию чужеродного гена. Если используется слишком сильный промотор, может возникнуть метаболическая «перегрузка», сдерживающая пролиферацию бактерий. В отличие от ферментера, организм животного-хозяина не является замкнутой системой, и экспрессию чужеродного гена нельзя регулировать изменением температуры или добавлением специфических метаболитов. Регуляторную роль может играть только промотор, реагирующий на те или иные сигналы. Например, работу промотора nirB E. coli можно регулировать, изменяя содержание нитритов и кислорода в среде, а наиболее активен он в анаэробных условиях. В одном из экспериментов промотор nirB использовали для контроля экспрессии гена нетоксичного иммуногенного С-фрагмента столбнячного токсина в аттенуированном штамме Salmonella. В развивающихся странах инфекция Clostridium tetani уносит более 1 млн. жизней в год. Если генетически модифицированный штамм Salmonella выращивать в аэробных условиях, то С-фрагмент столбнячного токсина синтезироваться не будет. При пероральном же введении этой бактерии тестируемым мышам С-фрагмент синтезируется, и у животных вырабатываются антитела к нему. Таким образом, штамм Salmonella, в который встроен С-фрагмент столбнячного токсина, находящийся под контролем промотора nirB, можно использовать как живую пероральную противостолбнячную вакцину. Чтобы выяснить, насколько эффективен этот подход при вакцинации человека, необходимо провести дополнительные исследования.
ЗАКЛЮЧЕНИЕ
Традиционные вакцины содержат инактивированные или аттенуированные патогенные микроорганизмы (бактерии или вирусы). Эти вакцины имеют ряд недостатков: не все патогенные микроорганизмы можно вырастить в необходимых для производства вакцины количествах; работа с большим количеством патогенных микроорганизмов требует соблюдения строжайших мер предосторожности; аттенуированные штаммы нередко ревертируют и становятся вирулентными; инактивация часто бывает неполной; срок годности вакцины зависит от условий ее хранения.
Технология рекомбинантных ДНКпозволяет создавать надежные вакцины, используя при этом разные подходы. Делетируя гены, ответственные за вирулентность, получают живые вакцины, содержащие непатогенные, иммунолога-чески активные штаммы, которые не могут
244 ГЛАВА 11
ревертировать и становиться патогенными. Клонированные гены, кодирующие основные антигенные детерминанты патогенного организма, встраивают в геном непатогенного носителя (обычно вируса) и получают безопасную, не содержащую болезнетворных микроорганизмов вакцину. Наконец, гены или их сегменты, кодирующие основные антигенные детерминанты патогенных микроорганизмов, встраивают в экспрессирующие векторы, получают нужный продукт в большом количестве и используют его как вакцину. Последний подход позволяет производить субъединичные и пептидные вакцины (если используются полноразмерные гены в первом случае и фрагменты генов, кодирующих домены основных антигенных детерминант — во втором). Пептидные вакцины получают и с помощью химического синтеза пептидов.
ЛИТЕРАТУРА
Andio R., D. Silvera, S. D. Suggett, P. L. Achacoso,
C. J. Miller, D. Baltimore, M. B. Feinberg.1994. Engineering poliovirus as a vaccine vector for the expression of diverse antigens. Science 265: 1448-1451.
Bittle J. L., R. A, Houghten, H. Alexander, T. M. Shinnick, J. G.Sutcliffc, R. A. Lernen,
D. J. Rowlands, F, Brown.1982. Protection against foot-and-mouth disease by immunization with a chemically synthesized peptide predicted from the viral nucleotide sequence. Nature 298: 30-33.
Blancou J., M. P. Kieny, R. Lathe, J. P.Lecoq, P. P. Pastoret, J. P. Soulebot, P. Desmettre.1986. Oral vaccination of the fox against rabies using a live recombinant vaccinia vaccine. Nature 322: 373-375.
Blasco R., B.Moss. 1995, Selection of recombinant vaccinia viruses on the basis of plaque formation. GeneW: 157-162.
Boothroyd J. C., P. E. Highfîeld, G. A,M. Cross, D. J. Rowlands, P. A. Lowe, F.Brown, T. J. R. Harris. 1981. Molecular cloning of foot and mouth disease virus genome and nucleotide sequences in the structural protein genes. Nature 290: 800-802.
Brown Г.1984. Synthetic viral vaccines. Annu. Rev. A/kro&O/. 38:221-235.
Brown F.1985. Peptides as the next generation of foot-and-mouth disease vaccines. Bio/Techno-logy 3:445-448.
Burnette W.N. 1990. The advent of recombinant pertussis vaccines. Bio/Technology 8: 1003-1005.
Charles L, G.Dougan. 1990. Gene expression and the development of live enteric vaccines. Trends Biotechnol. 8:117-121.
Chatfield S. N., I. G. Charles, A. J. Makoff, M. D. Oxer, G. Dougan, D. Pickard, D. Slater, N. F. Fairweather.1992. Use of the nirB promoter to direct the slnble expression of heterologous antigens in Salmonella oral vaccine strains: development of a single-dose oral tetanus vaccine. Bio/Technology 10:888-892.
Chow M., R. Yabrov, J. Bittle, J.Hogle, D. Baltimore.1985. Synthetic peptides from four separate regions of the poliovirus type 1 capsid protein VP1 induce neutralizing antibodies. Proc. Natl. Acad. Sei. USA 82: 910-914.
Clarke B. E.,S. E.Newton, A. R, Carroll, M. J. Francis, G. Appleyard, A. D.Syred, P. E. Highfield, D. J. Rowlands, F. Brown.1987. Improved immunogenicity of a peptide epitope after fusion to hepatitis В core protein. Nature 330: 381-384.
Cohen J.1993. Naked DNA points way to vaccines. Science 259: 1691-1692.
Cremer К. J., M.Macketl, С. Wohlenberg, A. L. Notkins, B.Moss. 1985. Vaccinia virus recombinant expressing herpes simplex virus type 1 glycoprotein D prevents latent herpes in mice. Science. 228: 737-740.
DiMarchi R., G. Brooke, C. Gale, V. Cracknel), T. Doel, N. Mowat.1986. Protection of cattle against foot-and-mouth disease by a synthetic peptide. Science 232: 639-641.
Ferguson M. 1991. Progress towards rabies control. Trends Biotechnol. 9: 7—11.
Finkelstein A., R. F. Silva.1989. Live recombinant vaccines for poultry. Trends Biotechnol. 7: 273-277.
Flexner C., A. Hugin, B.Moss. 1987. Prevention of vaccinia virus infection in immunodeficient mice by vector-directed IL-2 expression. Nature 330: 759 262
Вакцины 245
Graham F. L.1990. Adenoviruses as expression vector and recombinant vaccines. Trends Biotechnoi. 8: 85-87.
Horwitz M. A., B.-W. E. Lee, B. J. Dillon, G. Harth,1995. Protective immunity against tuberculosis induced by vaccination with major extracellular proteins of Mycobacterium tuberculosis. Proc. Natl. Acad. Sei. USA 92: 1530-1534.
Jones T. R., S. L. Hoffman. 1994. Malaria) vaccine development. Clin. Microbioi. Rev. 7: 303—310.
Kaper J. B., H. Lockman, M. M. Baldini,M. M. Levine. 1984. A recombinant live oral choiera vaccine. Bio/Technology 2: 345-349.
Kaper J. В., H. Lockman,M. M.Baldini, M. M. Levine. 1984. Recombinant nontoxinogenic Vibrio choier-ae strains as attenuated cholera vaccine candidates. Nature 308: 655-658.
Kaper J. В., J. G.Morris, Jr., M. M. Levine. 1995. Choiera. Clin. Microbioi. Rev. 8: 48-86.
Kaslow D. C., S. N. Isaacs L A. Quakyi, R. W. Gwadz, B,Moss, D. B. Keister.1991. Induction of Plasmodium falciparum transmission-blocking antibodies by recombinant vaccinia virus. Science 252: 1310-1313.
Кupper H., W. Keller, С. Kurz, S. Forss, H. Schauer, R. Franze, K. Strohmaier, O. Marquardt, V. G. Zaslavsky, P. H.Hofschneidtr. 1981. Cloning of cDNA of major antigen of fool and mouth disease virus and expression in E. colt. Nature 289: 555-559.
Lasky L. A., D. Dowbenko, C. C. Simonsen, P. W. Berman.1984. Protection of mice from lethal herpes simplex virus infection by vaccination with a secreted form of cloned glycoprotein D. Bio/Technology 2: 527-532.
Lowe R.S., P.M. Keller, B..!. Ketch, A. J. Uavison, Y. Whang, A. J.Morgan, E. KicfT, R. W. Elfe.1987. Varicella-zoster virus as a live vector for the expression of foreign genes, Proc. Natl. Acad. ScL USA 84: 3896-3900.
Mekalanos J. J., J. C. Sadoff.1994. Cholera vaccines: fighting an ancient scourge. Science 265: 1387-1389.
Michel M.-L., H. L. Davis, M. Schleef, M. Mancini, P.Tiollais, R. G. Whalen.1995. DNA-mediated immunization to the hepatitis В surface antigen in mice: aspects of the humoral response mimic hepatitis В viral infection in humans. Proc. Natl. Acad. Sei. USA 92: 5307-5311.
Miner J. N., D. E. Hruby.1990. Vaccinia virus: a versatile tool for molecular biologists. Trends Biotechnoi 8: 20-25.
Moss B. 1991- Vaccinia virus: a tool for research and vaccine development, Science252: 1662—1667.
Nabel G. J., P. L·Feigner. 1993. Direct gene transfer for immunotherapy and immunization. Tnends Biotechnoi. 11:211-215.
NewtonS. M. C., C.O. Jacob, B. A. U. Stocker. 1989. Immunoresponse to cholera toxin upitope inserted in Salmonella flagellin. Science 244: 70-72.
Nussenzweig R. S., C. A. Long.1994. Malaria vaccines: multiple targets. Science 265: 1381—1383.
Oehen S., H. Hengartner, R. M. Zinkernagel.1991. Vaccination for disease. Science 251: 195-197.
Paoletti E., J. TarlagHa January 1995. Interferon sensitive recombinant poxvirus vaccine. U.S. patent 5, 378,457.
Ratafia M. 1987. Worldwide opportunities in genetically engineered vaccines. Bio/Technology 5: N54-1158.
Sizemore D, R., A. A. Branstrom, J. C.Sadoff. 1995, Attenuated Shigella as a DNA delivery vehicle for DNA-mediated immunization. Science 270: 299-302.
Stover C. K., V. F. de la Cruz, T. R. Fuerst, J. E. Buriein, L· A. Benson, L.TBennett, G. P. Bansal, J. F. Young, M. H. Lee, G. F. Hatfull, S. B. Snapper, R. G. Barletta, W. R. Jacobs, Jr., B. R. Bloom.1991. New use of BCG for recombinant vaccines. Nature 351: 456—460.
Tang D.-C-, M. DeVit,S. A. Johnston. 1992. Genetic immunization is a simple method for eliciting an immune response. Nature 356: 152-154.
Tartaglia J., E. Paoletti.1988. Recombinant vaccinia virus vaccines. Trends Biotechnoi. 6: 43-46.
Titus R. GM J. G. Gueiros-Filtio, L. A. R. De Freitas, S. M. Beverly.1995. Development of a safe live Leishmania vaccine line by gene replacement. Proc. Natl. Acad. Sei. USA92: 10267-10271.
Uhner J. В., J. J.Donnelly, S. Parker, G. II. Rhodes, P. L. Feigner, V. J. Dwarki, S. H. Gromkowski, R. R. Deck, C. M, DeWïtt, A.Friedman, L. A. I lawe, K. R. Leander, D. Martine/, H. C. Perry, J. W. Shiver, D. I,. Montgomery, M. A. Liu. 1993.
246 ГЛАВА 11
Heterologous protection against influenza by injection of DN A encoding a viral protein. Science 259: 1745-1749.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Опишите вкратце способ создания вакцины против бактерий, продуцирующих токсин.
2. Что ограничивает применение традиционных вакцин?
3. Предположим, что вы принимаете участие в работе международной организации по охране здоровья животных и вам нужно создать вакцину против крайне вирулентного вируса крупного рогатого скота. Известно, что геном представляет собой полиаденилированную линейную одноцепочечную РНК длиной 10 т. п. н. и содержит восемь разных генов. Вирус не имеет оболочки, его основной антигенной детерминантой является белок капсида (VP 2). Какую стратегию вы используете?
4. Какие подходы применяются при создании пептидных противовирусных вакцин?
5. Что представляет собой вирус коровьей оспы и как с его помощью можно получать уникальные живые рекомбинантные вакцины?
6. Предположим, что вы выделили РНК-содержащий вирус, вызывающий бешенство у скунсов и енотов. Как на основе этого очищенного вируса создать рекомбинантную вакцину, защищающую животных от бешенства?
7. Как работник Всемирной организации здравоохранения вы должны найти оптимальный способ искоренения бешенства в популяциях диких животных. Предположим, что у вас есть пептидная вакцина и вакцина на основе вируса коровьей оспы; выберите одну из них и обоснуйте ваше решение.
8. Как можно использовать в качестве вакцины бактерии со жгутиками?
9. Перечислите преимущества живой рекомбинантной вирусной вакцины перед неживой и субъединичной вакцинами.
10.Опишите несколько подходов, использованных при создании холерной вакцины.
ГЛАВА 12
Использование рекомбинантных
микроорганизмов для получения
коммерческих продуктов
До настоящего времени основной целью исследований в области молекулярной биотехнологии было получение различных белков. Однако технологию рекомбинантных ДНК можно использовать также для крупномасштабного производства многих ценных низкомолекулярных соединений — витаминов, аминокислот, антибиотиков и т. д.
При наличии эффективной системы экспрессии получение белка — продукта специфического гена - не составляет особого труда. Белок может представлять собой либо тот конечный продукт, который хотят получить (например, рестрицирующую эндонуклеазу), либо фермент, катализирующий определенную химическую реакцию (например, одну из реакций биосинтеза антибиотиков). Иногда в результате генетических манипуляций микроорганизм приобретает способность к синтезу нового фермента и может использоваться для получения in vivo низкомолекулярных соединений — витаминов, аминокислот, красителей, антибиотиков, предшественников различных биополимеров и т. д. Такой микроорганизм становится «фабрикой» по производству полезных метаболитов.
Эндонуклеазы рестрикции
Развитие технологии рекомбинантных ДНК было бы невозможно, если бы в распоряжении исследователей не было нужных эндонуклеаз рестрикции (рестриктаз). В настоящее время в продаже имеется более 300 различных рестриктаз. Эти ферменты синтезируются самыми разными микроорганизмами: аэробными, анаэробными, фотосинтезирующими, диазотрофными, мезотрофными, термофильными, психрофильными, медленно- и быстрорастущими. Для культивирования каждого из них необходимо подобрать оптимальные условия ферментации температуру, pH, состав среды, концентрацию кислорода — с тем чтобы максимизировать выход необходимого фермента. Чтобы не пришлось выращивать большое число разных микроорганизмов, готовить многокомпонентные среды, разрабатывать разные ферментеры и тратить время на подбор оптимальных условий роста для многочисленных организмов, часто клонируют гены эндонуклеаз рестрикции в Escherichia coli. Это позволяет стандартизовать условия получения необходимых продуктов. Кроме того, культура клеток Е. coli быстро достигает высокой плотности и может быть приспособлена для сверхпродукции необходимого фермента.
Технология выделения и экспрессии чужеродных генов в E. coli и в некоторых других микроорганизмах достаточно хорошо отработана, однако не стоит забывать, что синтез гетерологичного белка в организме-хозяине может оказывать на него негативное влияние. Например, сверхпродукция такого белка может привести к истощению метаболических ресурсов хозяйского организма и отрицательно повлиять на его рост. Присутствие гетерологичного белка может оказаться даже губительным для клетки-хозяина. Так, сайты рестрикции имеются во всех молекулах ДНК, и если продуктом клонированного гена является эндонуклеаза рестрикции, то в отсутствие специальных зашитных механизмов хозяйская ДНК будет расщепляться ею.
248 ГЛАВА 12
Микроорганизмы, синтезирующие эндонуклеазы рестрикции, выработали систему самозащиты: они метилируют одно или несколько оснований рестриктазного сайта, и расщепление ДНК в этом сайте гомологичной эндонуклеазой рестрикции блокируется. Грамотрицательные микроорганизмы имеют еще один механизм защиты: эндонуклеазы рестрикции у них локализованы в периплазматическом пространстве. Благодаря такой компартментализации происходит физическое разделение рестриктаз и ДНК и при этом обеспечивается свободный доступ метилирующего (модифицирующего) фермента к хромосомной ДНК. Кроме того, это защищает клетку от проникновения в нее любой чужеродной ДНК, например вирусной.
Один из подходов к решению проблемы деградации хозяйской ДНК гетерологичными эндонуклеазами рестрикции состоит в клонировании и экспрессии в реципиентном организме как гена фермента рестрикции, так и гена соответствующего модифицирующего фермента. Однако клонирование обоих этих генов в одном микроорганизме технически затруднено, если они расположены на хромосоме донорного организма далеко друг от друга. Кроме того, чтобы не допустить расщепления хозяйской ДНК эндонуклеазами рестрикции, метилирующий фермент после трансформации должен синтезироваться еще до начала синтеза рестриктазы.
На рис. 12.1 представлена стратегия выделения и клонирования в Е. coli гена рестриктазы Pstl грамотрицательной бактерии Providencia stuartii
1. ДНК P, stuartii расщепляют с помощью HindIIIи встраивают фрагменты в HindIII-сайт плазмиды pBR322.
2. Рекомбинантными плазмидами трансформируют клетки E. соli НВ101 и выращивают их в жидкой среде, а затем инфицируют бактериофагом λ. Если в хозяйской клетке экспрессируется ген фермента рестрикции, то она оказывается устойчивой к литическому действию фагов типа λ, ДНК которых активно расщепляется синтезируемой рестриктазой.
3. Трансформированные клетки, устойчивые к фагу λ, подвергают осмотическому шоку, чтобы высвободить периплазматические белки. Определяют активность рестриктазы PstI в белковом экстракте.
4. Положительные клоны тестируют на наличие PstI-метилирующей активности.
Один положительный клон, выявленный в этом эксперименте, содержал встроенный фрагмент ДНК длиной 4 т. п. н. с интактным опероном рестриктазы и метилазы Art и промотором P. stuartii В клоне, несущем эту генетическую конструкцию, соблюдался естественный временной порядок синтеза: вначале синтезировался метилирующий фермент, затем эндонуклеаза рестрикции. Уровень экспрессии гена рестриктазы PsA в Е. colt был примерно в 10 раз выше, чем в P. stuartii. Как и предполагалось, рестриктаза находилась в периплазматическом пространстве, а метилаза — в цитоплазме. Метод получения PstI клонированием соответствующего гена в Е. соli гораздо более эффективен, чем выделение этого фермента из P. stuartii.
Для выделения генов, кодирующих ферменты рестрикции и модификации (метилирования), можно использовать также другой подход, который состоит в следующем,
1. Создают банк клонов ДНК организма-донора, продуцирующего известную эндонуклеазу рестрикции. Используемый при этом плазмидный вектор должен содержать по крайней мере один сайт узнавания для этой рестриктазы.
2. Трансформируют Е. colt гибридными плазмидами.
3. Из трансформированных клеток, выросших в жидкой селективной среде (т. е. из клеток, содержащих плазмиду), выделяют плазмиднуюДНК.
4. Обрабатывают ее интересующей исследователя эндонуклеазой рестрикции,
5. Трансформируют Е. coli плазмидными ДНК, обработанными эндонуклеазой рестрикции.
Ключевым моментом этого метода является то, что плазмидная ДНК клонов, несущих и экспрессирующих ген фермента модификации, оказывается устойчивой к расщеплению соответствующей эндонуклеазой рестрикции, поскольку сайты узнавания в ней метилированы.
Использование рекомбинантных микроорганизмов для получения коммерческих продуктов 249
| Рис. 12.1. Клонирование гена рестриктазы PstI и отбор несущих его трансформированных бактериальных клеток. Хромосомную ДНК P. stuartii расщепляют HindIIIи встраивают фрагменты в плазмиду pBR322. Трансформируют рекомбинантной плазмидой Е. coli, выращивают клетки в жидкой среде и инфицируют фагом λ. Отбирают трансформанты, устойчивые к фагу; именно они несут и э кспрессируют клонированный ген PstI. | 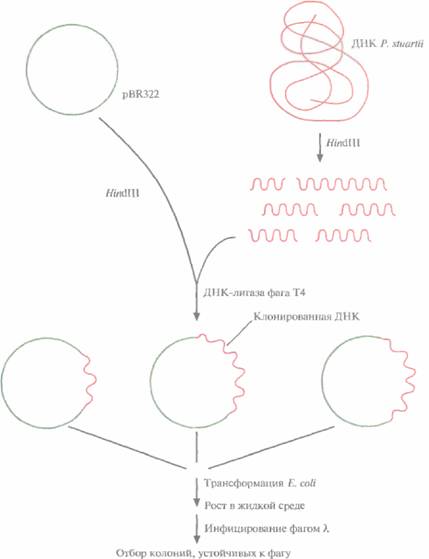
|
Рассмотрим следующий пример. В плазмиду pBR322 встраивали HindIII-фрагменты ДНК Desulfovibrio desulfuricans и трансформировали ею клетки Е. coli. Выделенную из трансформированных клеток плазмидную ДНК обрабатывали рестриктазой DdeI. Плазмиды, несущие и экспрессирующие ген метилирующего фермента, не расщеплялись, поскольку все восемь сайтов узнавания DdeIв pBR322 были метилированы. Смесь плазмид, обработанных Ddel, использовали для трансформации E. coli. Образование трансформантов, несущих ген функционального модифицирующего фермента DdeI, обеспечивали только целые кольцевые молекулы плазмидных ДНК. Остальные плазмиды были расщеплены эндонуклеазой рестрикции. Для того чтобы определить, какие клоны содержат и ген фермента модификации, и ген эндонуклеазы рестрикции, трансформанты тестировали на наличие в них активной рестриктазы DdeI. Описанный подход можно с успехом использовать для выделения гена любой рестриктазы, лишь бы он находился достаточно близко к гену соответствующего модифицирующего фермента и
250 ГЛАВА 12
был встроен в плазмидный вектор, имеющий по меньшей мере один сайт узнавания для данного фермента.
Малые биологические молекулы
Используя технологию рекомбинантных ДНК, можно направленно изменять метаболизм микроорганизмов, вводя в них новые гены или модифицируя уже существующие. Основной целью таких изменений является создание рекомбинантного микроорганизма с новой ферментативной активностью, способного превращать существующий субстрат в ценный продукт, который обычно получают только сочетанием химических и микробиологических методов.
Синтез L-аскорбиновой кислоты
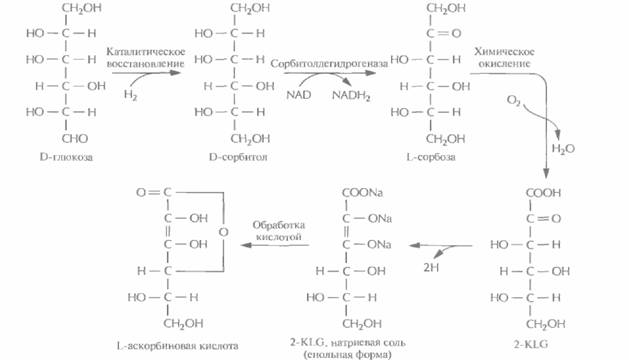
В настоящее время для крупномасштабного производства L-аскорбиновой кислоты (витамина С) используют весьма трудоемкий процесс, включающий одну микробиологическую стадию и несколько химических; исходным субстратом для него является D-глюкоза (рис. 12.2). На последнем этапе этого процесса 2-кето-L-гулоновая кислота (2-KLG) превращается в кислых условиях в L-аскорбиновую кислоту. Биохимические исследования метаболизма различных микроорганизмов показали, что 2-KLG можно получить другим путем. Так, одни бактерии (Acetobacter, Gluconobacter и Erwinia) могут превращать глюкозу в 2,5-дикето-О-глюконовую кислоту (2,5-DKG), а другие (Corynebacterium, Brevibacterium и Arthrobacter), синтезирующие фермент 2,5-DKG-редуктазу, — преобразовывать 2,5-DKG в 2-KLG.
Использующийся в настоящее время способ получения аскорбиновой кислоты можно усовершенствовать, если включить в него совместное культивирование указанных микроорганизмов для превращения глюкозы в 2-KLG. К сожалению, такое культивирование имеет свои трудности. Например, используемые микроорганизмы могут иметь разные оптимумы температуры и pH, могут различаться также состав среды и скорость роста. Иными словами, условия культивирования, оптимальные для одного организма, могут быть неприемлемы для другого, что приведет к спонтанному «вымыванию" из среды одного из них. В подобных случаях можно культивировать микроорганизмы последовательно (рис. 12.3), правда такой процесс трудно будет сделать непрерывным, если для роста микроорганизмов необходимы существенно разные среды. Наилучшим выходом из этой ситуации было бы создание одного микроорганизма, синтезирующего все ферменты, необходимые для превращения глюкозы в 2-KLG. Erwinia herbicola осуществляет превращение D-глюкозы в 2,5-DKG в несколько стадий, катализируемых разными ферментами, в то время как Corynebacterium sp. для превращения 2,5-DKG в 2-KLG необходима только одна стадия. Следовательно, наиболее простой способ создания одного микроорганизма, способного превращать D-глюкозу в 2-KLG, состоит в выделении гена 2,5-ОКО-редуктазы Coiynebacterium sp. и введении его в Erwmia herbicoia.
Первый шаг на этом пути состоит в выделении и очистке 2,5-DКG-редуктазы Coiynebacterium sp. и определении последовательности ее первых 40 N-концевых аминокислот. Исходя из этих данных были синтезированы два 43-нуклеотидных гибридизационных зонда, соответствовавших разным частям белковой молекулы. Поскольку 71% нуклеотидов ДНК Corynebacterium sp. представляют собой либо G, либо С, зонды синтезировали таким образом, чтобы в третьем положении кодонов по возможности находились именно они. Это позволяло минимизировать число неспаренных оснований между зондами и искомой ДНК.
Синтезированные зонды использовали для скрининга банка клонов ДНК Corynebacterium·, клоны, гибридизующиеся только с одним из зондов, исключали из дальнейшего рассмотрения, считая, что соответствующая ДНК не является искомой. Выделяли клон, содержащий ген 2,5-DКG-редуктазы, и секвенировали его. Нуклеотидные последовательности, расположенные до стартового кодона ATG, вырезали и заменяли их сигналами транскрипции и трансляции, функционирующими в Е. coli, поскольку регуляторные последовательности грамположительных микроорганизмов типа Corynebacterium spp. не функционируют в клетках этого микроорганизма. Полученную конструкцию вводили в
Использование рекомбинантных микроорганизмов для получения коммерческих продуктов 251
|
| Рис. 12.2. Промышленный синтез L-аскорбиновой кислоты. Одна из стадий процесса, а именно превращение D-сорбитола в L-сорбозу, осуществляется при участии бактерии Acetobacter suboxydans, которая синтезирует фермент сорбитолдегидрогеназу. Остальные стадии представляют собой чисто химические реакции. |
Е. coli (при этом синтезировалась активная 2,5-DKG-редуктаза), а затем переклонировали в векторе с широким кругом хозяев и трансформировали им Erwinia herbicola.
Трансформированные клетки Erwinia активно превращали D-глюкозу непосредственно в 2-KLG, при этом собственные ферменты Erwinia, локализованные во внутренней мембране бактериальной клетки, преобразовывали глюкозу в 2,5-DKG, а 2,5-DКG-редуктаза, локализованная в цитоплазме, катализировала превращение 2,5-DKG в 2-KLG (рис. 12.4). Таким образом, с помощью генетических манипуляций метаболические реакции, протекающие в столь разных микроорганизмах, удалось осуществить в одном из них. Этот гибрид приобрел способность синтезировать конечный продукт комбинированного метаболического пути. Такой организм можно использовать как фабрику для производства 2-KLG, заменяющую первые три стадии в том процессе получения L-аскорбиновой кислоты, который используется в настоящее время (рис. 12.2).
Коммерческую ценность 2,5-DKG-редуктазы можно повысить, если произвести аминокислотные замены, повышающие каталитическую активность фермента и его термостабильность. В то время, коша был идентифицирован ген 2.5-DKG-редуктазы, аминокислотные остатки, участвующие в образовании активного центра этого фермента, еще не были установлены. Однако, исходя из данных об аминокислотной последовательности фермента, была воссоздана его вторичная структура, состоящая из восьми тесно расположенных параллельных β-слоев, перемежающихся восемью α-спиралями, которые соединялись с β-слоями петлями разной длины (рис. 12.5). Такой характер укладки полипептидной цепи был установлен для 17 других ферментов с уже известной кристаллической структурой, и по аналогии с ними были идентифицированы три петли, возможно участвующие в связывании субстрата. С помо-
252 ГЛАВА 12

| Рис. 12.3. Микробиологический синтез 2-KLG. Erwinia продуцирует три фермента, обеспечивающих синтез 2,5-DKG из D-глюкозы, a Corynebacteriun - фермент, катализирующий превращение 2,5-DKG e 2-KLG. Таким образом, 2-KLG, непосредственный предшественник L- аскорбиновой кислоты, можно синтезировать из D-глюкозы совместным культивированием этих двух микроорганизмов. Альтернативный подход состоит в создании рекомбинантной бактерии Erwinia, экспрессирующей ген соответствующего фермента Corynebaclerium. |
щью олигонуклеотид-направленного мутагенеза были получены 12 мутантных белков, каждый из которых содержал одну аминокислотную замену в одной из петель. 11 мутантных форм 2,5-DKG-редуктазы обладали более низкой удельной активностью, чем нативный фермент, а 12-я, у которой остаток глутамина в положении 192 был заменен на аргинин, была примерно в два раза более активной. По данным кинетических исследований, повышение активности было связано с увеличением в 1,8 раз максимальной скорости (Vmax) и уменьшением на 25% константы Михаэлиса (КM) реакции, катализируемой ферментом. Замена глициновых остатков в положениях 55 и 57 на аланиновые позволила получить более термостабильный фермент по сравнению с нативной формой. Дальнейшие усилия будут, вероятно, направлены на получение фермента, сочетающего оба этих свойства.
Синтез индиго
Множество бактерий, особенно бактерии вида Pseudomonas, способны утилизировать различные органические соединения типа нафталина, толуола, ксилола и фенола, которые являются для них единственным источником углерода. Очень часто гены ферментов, катализирующих расщепление этих органических соединений, располагаются в крупных природных плазми-дах (длиной 50—200 т.п.н.). Чтобы ставить эксперименты с этими бактериями, в частности проводить целенаправленную модификацию генов ферментов, катализирующих те или иные метаболические реакции, приходится предпринимать детальные генетические и биохимические исследования, и нередко в ходе этих исследований делаются неожиданные и весьма интересные открытия. Рассмотрим следующий пример. Плазмида NAH7 содержит два разных оперона, которые позволяют несущим ее псевдомонадам использовать нафталин как единственный источник углерода. Для характеристики соответствующих генов расщепили плазмидную ДНК с помощью HindIII и лигировали фрагменты с линеаризованной HindIII плазмидой pBR322. Полученные гибридные молекулы ввели в клетки Е. соli и отобрали трансформантов, устойчивых к ампициллину, но чувствительных к тетрациклину. Затем проверили всех трансформантов на способность образовывать нелетучие метаболиты — возможные продукты гидролиза радиоактивно меченного нафталина.
При исследовании одного из трансформантов, содержащего вставку длиной 10,5 т.п.н. и способного превращать нафталин в салициловую кислоту, обнаружилось, что минимальная ростовая среда, содержащая триптофан, приобретает синюю окраску. Тщательный анализ этого явления показал, что трансформированные клетки E. coli синтезировали краситель индиго. Синтез происходил в четыре стадии (рис. 12.6).
Использование рекомбинантных микроорганизмов для получения коммерческих продуктов 253
| Рис. 12.4. Превращение D-глюкозы в 2-KLG рекомбинантной бактерией Erwinia herbicola. Все участвующие в этом процессеферменты обозначены буквой Ε и последовательно пронумерованы, указана также их клеточная локализация. | 
|
| Рис. 12.5. Структура 2,5-DKG-peдуктазы, воссозданная исходя из ее аминокислотной последовательности. Стрелки — β-слои, полоски -α-спиральныеучастки, кружки аминокислотные остатки, находящиеся на N- и С-концах молекулы или соединяющие ß-слои и α-спирали. Показаны три петли, возможно, участвующие в связывании субстрата. Желтый кружок — 192-й аминокислотный остаток. | 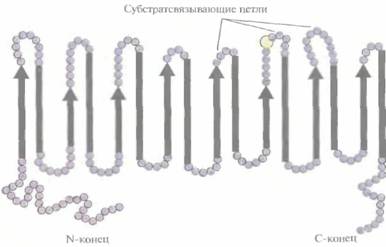
|
Дата добавления: 2015-07-14; просмотров: 1106;