ПЕРЕГОНКА ТА РЕКТИФІКАЦІЯ
3.1. Загальні відомості
Одним з найпоширеніших методів розділення рідких однорідних сумішей, що складаються з двох або більшої кількості летких компонентів, є перегонка (дистиляція і ректифікація).
Перегонка – це процес, що передбачає часткове випаровування суміші, що розділяється, і подальшу конденсацію пари, що утворюється. Може здійснюватися одноразово або багато разів. У результаті конденсації одержують рідину, склад якої відрізняється від складу початкової суміші.
Якщо би початкова суміш складалася з леткого або нелеткого компонентів, то її можна було б розділити на компоненти шляхом випарювання. Перегонкою розділяють суміші, всі компоненти яких леткі, тобто мають значний, хоча і різний тиск пари. Розділення перегонкою засновано на різній леткості компонентів суміші за однакової температури. Тому при перегонці всі компоненти суміші переходять в пароподібний стан в кількостях, пропорційних їх фугітивності (леткості).
У спрощеному випадку початкова суміш є бінарною, тобто складається тільки з двох компонентів. Отримана під час перегонки пара містить відносно більшу кількість легколеткого (ЛЛК) або низькокиплячого (НКК) компоненту, ніж початкова суміш. Отже, під час перегонки рідка фаза збіднюється, а парова фаза збагачується ЛЛК. Рідина, що не випарувалася, має склад, багатший на важколеткий (ВЛК), або висококиплячий(ВКК) компоненти.
Ця рідина називається залишком, а рідина, отримана в результаті конденсації пари – дистилятом, або ректифікатом.
Ступінь збагачення парової фази ЛЛК за інших рівних умов залежить від виду перегонки. Існують два принципово відмінні види перегонки: 1) проста перегонка (дистиляція) і 2) ректифікація.
Проста перегонка є процесом одноразового часткового випаровування рідкої суміші і конденсації пари, що утворюється. Проста перегонка застосовується тільки для розділення сумішей, леткість компонентів якої істотно відрізняється, тобто відношення леткості (відносна леткість) компонентів значна. Ії використовують лише для попереднього грубого розділення рідких сумішей, а також для очищення складних сумішей від небажаних домішок, смол і т.п. Відомі декілька різновидів простої перегонки, що розглядатимуться нижче.
Повніше розділення рідких сумішей на компоненти досягається шляхом ректифікації.
Ректифікація –процес розділення гомогенних сумішей летких рідин двостороннім масо- і теплообміном між нерівноважними рідкою і паровою фазами, що мають різну температуру і рухаються назустріч один одному. Розділення здійснюється в колонних апаратах при багатократному або безперервному контакті фаз. При кожному контакті з рідини випаровується переважно ЛЛК, яким збагачується пара, а з парової фази конденсується переважно ВЛК, що переходить в рідину. Обмін компонентами між фазами дає змогу отримати пару, яка є майже чистим ЛЛК. Ця пара, що виходить з верхньої частини колони, після конденсації в окремому апараті розділяється на дистилят, або ректифікат (верхній продукт) і флегму – рідину, що повертається для зрошування колони і взаємодії з парою, яка підіймається по колоні. Знизу колони відділяється рідина, що є майже чистим ВЛК, – залишок (нижній продукт). Частину залишку випаровують в нижній частині колони для отримання висхідного потоку пари.
Ректифікація відома з початку XIX століття як один з найважливіших технологічних процесів переважно спиртової і нафтової промисловості. Сьогодні ректифікацію все ширше застосовують у різноманітних областях хімічної технології, де виділення компонентів у чистому вигляді має вельми важливе значення (у виробництвах органічного синтезу, ізотопів, полімерів, напівпровідників та інших речовин високої чистоти).
Процеси перегонки здійснюються періодично або безперервно. Нижче розглянуті характеристики двофазних систем “рідина – пара”, необхідні для аналізу і розрахунку процесів перегонки.
3.2. Характеристики двофазних систем “рідина – пара”
Фазова рівновага бінарних сумішей. Якщо система складається з двох компонентів (К = 2) і між ними не відбувається хімічної взаємодії, то за наявності рідкої і парової фаз кількість фаз Ф = 2. За правилом фаз кількість ступенів вільності такої системи становить
С = К +2 – Ф = 2 + 2 – 2=2.
Отже, з трьох незалежних параметрів, які повністю визначають стан системи, – температура t, тиск р і концентрація однієі з фаз с – можна довільно вибрати будь-яких два; при цьому визначиться значення третього параметра, яке вже не може бути довільним.
У зв’язку з цим для фізико-хімічної характеристики бінарних систем “рідина – пара” зручно користуватися так званими фазовими діаграмами. Якщо позначити через х склад рідкої фази, а через у – склад парової фази, то, приймаючи t = const, можна побудувати графік залежності тиску пари від складу рідини (діаграма р – х).’
Приймаючи сталим тиск над сумішшю (р = const), зображають на площині залежність температур кипіння рідини і концентрації пари від складів рідкої і парової фаз (діаграма t – х – у). Так самоза р = const і t = const знаходятьзалежність між рівноважними складами фаз, яка зображається діаграмою рівноваги (діаграма у – х).
Вид цієї залежності визначається взаємною розчинністю компонентів рідкої суміші та іншими їхніми властивостями, що розглядатимуться нижче.
Класифікація бінарних сумішей.Залежно від взаємної розчинності компонентів розрізняють суміші рідин: 1) з необмеженою взаємною розчинністю; 2) взаємно нерозчинних; 3) обмежено розчинних одна в одній. Суміші з необмеженою взаємною розчинністю компонентів своєю чергою поділяються на ідеальні і реальні (неідеальні) суміші.
Суміші рідин з необмеженою взаємною розчинністю
Ідеальні суміші. Ідеальні розчини відповідають закону Рауля. Згідно з законом Рауля, парціальний тиск кожного компонента, наприклад, низькокиплячого компонента А в парі рА, є пропорційним мольній частці хА цього компонента в рідині. При цьому коефіцієнт пропорційності дорівнює тиску насиченої пари РА цього компонента за певної температури.
У разі змішування компонентів ідеального розчину тепловий ефект є відсутнім і об’єм суміші практично не змінюється.
У загальному вигляді закон Рауля формулюється так: леткість fi будь-якого компонента ідеального розчину дорівнює леткості fio чистого компонента, помноженій на його мольну частку:
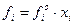 , (3.1)
, (3.1)
причому 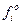 за заданих температури і загального тиску є величиною сталою, а відношення
за заданих температури і загального тиску є величиною сталою, а відношення 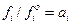 , тобто дорівнює активності розчину.
, тобто дорівнює активності розчину.
Для бінарної суміші, що складається з компонентів А і В, за законом Рауля
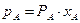 ,
,
 ,
,
де хВ – мольна частка компонента В, тиск насиченої пари якого над чистою рідиною за цієї температури дорівнює РВ.
Водночас, згідно з законом Дальтона, загальний тиск пари над розчином Р дорівнює сумі парціальних тисків його компонентів
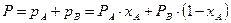 . (3.2)
. (3.2)
З рівнянь, що виражають закони Рауля і Дальтона, видно, що за сталої температури парціальні тиски компонентів, а також загальний тиск пари над сумішшю знаходяться в лінійній залежності від мольної частки хА низькокиплячого компонента.
На рис. 3.1 за t = const прямі ОВ і СА зображають зміни парціальних тисків компонентів (рА і рВ), а пряма АВ – зміну загального тиску над розчином. Вертикальні відрізки СВ і ОА виражають тиск насиченої пари чистих компонентів (РА і РВ).
Температура кипіння суміші заданого складу (хА) є функцією тиску пари. Для того, щоб її визначити, будують за значеннями тисків насиченої пари чистих компонентів (з довідників) ізотерми АВ, 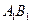 ,
, 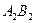 і т. д., виражаючи загальний тиск пари за температур
і т. д., виражаючи загальний тиск пари за температур 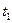 ,
, 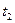 ,
, 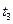 і т.д. Лінії парціальних тисків рА і рВ одержують, сполучаючи прямими крапки А,
і т.д. Лінії парціальних тисків рА і рВ одержують, сполучаючи прямими крапки А, 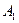 ,
, 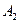 і т.д. з точкою С і точки В,
і т.д. з точкою С і точки В,  ,
,  і т.д. –з точкою О.
і т.д. –з точкою О.
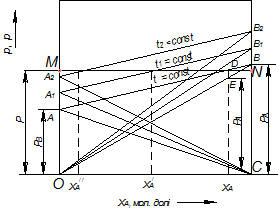
Рис. 3.1. Ізотерми парціальних тисків компонентів і загального тиску
для ідеальних розчинів (діаграма р – х)
Далі проводять горизонтальну пряму MN відповідно до зовнішнього тиску. З точки на осі абсцис, які відповідають хА, відновлюють вертикаль до перетину з прямою MN в точці D, через яку проходить ізотерма t = const. За цієї температури загальний тиск пари суміші дорівнює зовнішньому тиску, і, отже, суміш складу хА кипить за температури t. За аналогією можна визначити, що температура кипіння суміші складу х/А дорівнює суміші складу х//А – t2 і т.д.
Парціальний тиск рА низькокиплячого компонента над сумішшю за температури кипіння t зображається вертикальним відрізком, отриманим в результаті проведення вертикалі з точки, що відповідає суміші складу хА, до перетину з прямою парціальних тиску в точці Е.
Водночас відповідно до закону Дальтона (за умови застосовності до пари кожного з компонентів рівняння Менделєєва–Клапейрона), парціальний тиск рА заданого компонента А пропорційний до його мольної частки уА в парі:
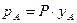 ,
,
де Р – загальний тиск пари над сумішшю.
Враховуючи, що за законом Рауля рА = Ра×xА, знайдемо вираз для складу пари у*А, що є рівноважною з рідиною заданого складу хА, тобто знайдемо залежність між складами рівноважних фаз:
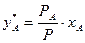 . (3.3)
. (3.3)
Для побудови залежності температур кипіння і конденсації від складу рідини або пари, тобто діаграми t – х – у (рис. 3.2), відкладають на осі ординат за сталого зовнішнього тиску, температури кипіння t1, t2, t3, що відповідаютьскладам рідких сумішей х1, х2, х3, відкладенимна осі абсцис. Через отримані точки, що відповідають температурам кипіння чистих компонентів tA і tВ, відкладених на крайніх ординатах діаграми, проводять лінію кипіння АА1А2А3В. Потім на осі абсцис відкладають визначені за законом Рауля рівноважні склади пари 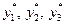 і проводять з відповідних їм точок прямі до перетину з ізотермами, що відповідають температурам t1, t2, t3. З’єднавши точки перетину В1, B2, B3, з точками А і В плавною кривою, одержують лінію конденсації А В1 В2 В3 В.
і проводять з відповідних їм точок прямі до перетину з ізотермами, що відповідають температурам t1, t2, t3. З’єднавши точки перетину В1, B2, B3, з точками А і В плавною кривою, одержують лінію конденсації А В1 В2 В3 В.
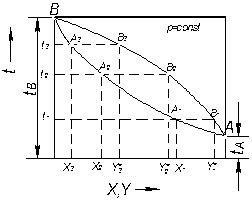
Рис. 3.2. Залежність температур кипіння і конденсації
від складу фаз (діаграма t – х – у)
У разі користуванні діаграмою на осі абсцис відкладають склад рідкої суміші і проводять з відповідної крапки вертикаль до перетину з лінією кипіння. Далі з точки перетину проводять горизонталь праворуч до перетину з лінією конденсації. Абсциса точки перетину вказує склад рівноважної пари.
На фазову діаграму у – х (рис. 3.3) наносять лінію рівноваги, що виражає залежність між рівноважними складами (за низькокиплячим компонентом) рідкої ( 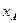 ) і парової
) і парової 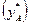 фаз. Процеси перегонки здійснюють зазвичай при сталому зовнішньому тиску. Тому діаграму будують за Р = const, тобтодля змінних температур кипіння, що змінюються залежно від зміни складу рідкої суміші.
фаз. Процеси перегонки здійснюють зазвичай при сталому зовнішньому тиску. Тому діаграму будують за Р = const, тобтодля змінних температур кипіння, що змінюються залежно від зміни складу рідкої суміші.
З рідиною, що є чистим НК, знаходиться в рівновазі пара, в якій міститься 100 % того самого компонента. Відповідно, крайні точки кривої рівноваги розташовані у протилежних кутках квадрата (рис. 3.3). Опуклість кривої рівноваги щодо діагоналі квадрата загалом залежить від відношення теплот випаровування компонентів суміші rA/rB. За rA/rB <1 із збільшенням тиску крива рівноваги стає менш опуклою, тобто, наближається до діагоналі квадрата (пунктир на рис. 3.3).
Для ідеальних розчинів рівняння лінії рівноваги 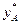 =f(х) можна вивести аналітично на основі законів Рауля і Дальтона. Підставивляючи значення Р з рівняння (3.2) в рівняння рівноваги (3.3) і розділивши чисельник і знаменник на
=f(х) можна вивести аналітично на основі законів Рауля і Дальтона. Підставивляючи значення Р з рівняння (3.2) в рівняння рівноваги (3.3) і розділивши чисельник і знаменник на 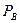 , отримаємо
, отримаємо
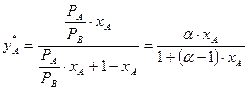 , (3.4)
, (3.4)
де a = Ра/Рв – відносна леткість компонентів суміші, дорівнює відношенню тисків пари чистих компонентів за умов однакового зовнішнього тиску.
Отже, знаючи a, тобто тиск насиченої пари чистих компонентів, можна розрахувати і побудувати криву рівноваги для ідеальних сумішей.
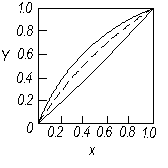
Рис. 3.3. Діаграма рівноваги “пара – рідина”
(діаграма у – х)
Насправді ідеальних розчинів не існує, проте в промисловості доводиться розділяти перегонкою багато розчинів, що наближаються за властивостями до ідеальних. До них належать подібні за хімічними властивостями речовини, наприклад, бензол і толуол, для яких побудовано діаграму у – х на рис.3.3.
Взаємне розташування кривих на фазових діаграмах t – х – у і у – х визначається першим законом Коновалова: пара збагатчується тим компонентом, при додаванні якого до рідини підвищується тиск пари над нею або знижується температура кипіння рідини.
Як видно з рис. 3.2, за однієї температури вміст НК в парі (точка В) більше його вмісту в рівноважній з парами рідині (точка А), тобто пару збагатили НК. Подібний висновок можна зробити, аналізуючи рис. 3.3. Крива рівноваги опукла догори. Додавання НК (на прикладі бензолу) до суміші бензол – толуол призводить до зменшення температури її кипіння. Відповідно концентрація бензолу в парі є вищою, ніж в рідині.
Перший закон Коновалова доповнюється першим законом Вревського, що визначає, як впливає температура (і відповідно тиск) на рівноважні склади фаз: за підвищення температури розчину двох рідин в парі зростає відносний вміст того компоненту, перехід якого в пароподібну фазу вимагає найбільшої витрати тепла.
Обидва закони (Коновалова і Вревського) є наслідком загального принципу зсуву рівноваги, сформульованого Ле-Шательє: якщо на систему, що знаходиться в стані стійкої рівноваги, подіяти, змінюючи умови, що визначають стан рівноваги, то відбудеться зсув рівноваги в тому напрямку, за якого ефект зовнішньої дії зменшується.
Для сумішей двох рідин (двокомпонентна система) такими зовнішніми діями можуть бути зміни температури, тиску і концентрації.
Реальні суміші. Реальні рідкі суміші з повною взаємною розчинністю компонентів не відповідають закону Рауля. Відхилення від цього закону в кожній з фаз може бути позитивним або негативним, причому останнє спостерігається рідше. У разі позитивного відхилення від закону Рауля різниця тиску Р – Рід > 0, при негативному відхиленні Р – Рід < 0, де Р – загальний тиск над реальним розчином, а Рід – над ідеальним розчином.
Відхилення від закону Рауля пов’язані із зміною активності молекул в розчині, обумовленою хімічною взаємодією між ними, дисоціацією, гідратацією (у водних розчинах) та ін. Ступінь відхилення властивостей реального розчину від властивостей ідеального розчину визначається величиною коефіцієнта активності g, що дорівнює відношенню активності компоненту розчину до його концентрації. На відміну від ідеального розчину, для якого gА = gВ = 1, парціальний тиск компонентів А і В внеідеальній бінарній суміші становить
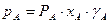 ;
; 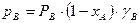 .
.
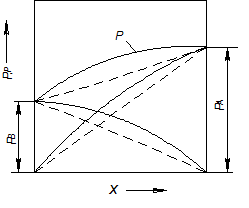
Рис. 3.4. Діаграма р – х для суміші з позитивним відхиленням
від закону Рауля (пунктиром показані відповідні лінії для ідеального розчину)
Для сумішей з позитивним відхиленням від закону Рауля g > 1, і лінія загального тиску над сумішшю проходить вище від прямої для ідеального розчину (рис. 3.4). Відповідно зміни парціального тиску компонентів також зображаються опуклими кривими.
Для сумішей з негативним відхиленням від закону Рауля g < 1, і лінії парціальних тисків компонентів, а також лінія загального тиску є увігнутими кривими.
Для неідеальних сумішей крива рівноваги може бути розрахована тільки за відомими коефіцієнтами активності, визначення яких є складним. Тому діаграми
у – х для реальних розчинів зазвичай будують на основі дослідних даних.
Для багатьох сумішей кількісні відхилення від закону Рауля є настільки великими, що приводять до якісно нових властивостей сумішей. За деякого складу подібні суміші мають сталу температуру кипіння, яка може бути максимальною або мінімальною. За цієї температури, згідно з загальним законом Коновалова, склад рівноважної пари над сумішшю дорівнює складу рідини (у=х). Такі суміші називають азеотропними, або нероздільно киплячими.
Типові діаграми р – х, t – х – у і у – х для систем з максимумом температури кипіння показані на рис. 3.5, а і з мінімумом температури кипіння – на рис.3.5, б. Як видно з рис.3.5, максимуму загального тиску пари відповідає мінімум температури кипіння суміші і навпаки.
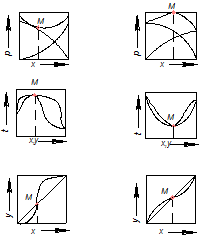
а б
Рис.3.5. Фазові діаграми азеотропних сумішей:
а – з максимумом температури кипіння; б – з мінімумом температури кипіння
Точка М, в якій криві складів фаз перетинають діагональ на діаграмі х – у, називається азеотропною точкою. Вона вказує склад суміші, яка за заданого тиску не може бути розділена перегонкою на складові компоненти.
Склад азеотропних сумішей залежить від температури (тиску). Ця залежність характеризується другим законом Вревського: у разі підвищення температури азеотропної суміші, що має максимум тиску пари, в суміші збільшується відносний вміст того компонента, парціальна мольна теплота випаровування якого є більшою, а для суміші з мінімумом тиску пари – вміст компонента, парціальна мольна теплота випаровування якого менша.
Цей закон вказує на принципову можливість розділення азеотропних сумішей внаслідок зміни тиску під час перегонки. Із зміною тиску азеотропна точка переміщається рівноважною кривою. У разі збігу цієї точки з правим верхнім кутом квадрата діаграми у – х зазвичай вдається розділити азеотропну суміш перегонкою.
Іноді крива рівноваги майже торкається горизонтальної прямої, проведеної через точку М (рис.3.5, а); тоді вказаній точці також відповідає азеотропна суміш (так званий тангенціальний азеотроп), і розділити початковий розчин неможливо.
Відомо багато нероздільно киплячих бінарних сумішей, що складаються з рідин, які є обмежено взаємно розчинними (рис. 3.6).Такі системи, при ректифікації яких одержуваний дистилят має той самий склад, що і пара, рівноважна з кожною з двох існуючих фаз, називаються гетерогенними азеотропними сумішами, або гетероазеотропами.
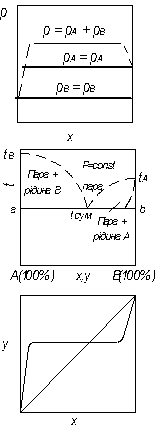
Рис. 3.6. Фазові діаграми сумішей взаємно нерозчинних рідин
Перехід гетероазеотропів в звичайні азеотропні суміші (гомоазеотропи) відбувається за достатньо високого тиску, коли температура кипіння суміші стає вищою за критичну точку розчинності компонентів.
Як буде показано нижче, розділення азеотропних сумішей здійснюють також за допомогою спеціальних видів перегонки, здійснюваних введенням в систему додаткового компонента.
Закони Коновалова і Вревського для систем “пара – рідина” належать до законів, що є основою процесів перегонки.
Суміші взаємно нерозчинних рідин
Практично взаємно нерозчинними вважаються рідини, що мають низьку розчинність одна в одній, якою нехтуютть. Прикладом може бути система “вода – бензол” та інші системи, що складаються з води і різних органічних речовин. Такі суміші утворюють два шари і можуть бути розділені відстоюванням.
Система, що складається з двох взаємно нерозчинних компонентів і трьох фаз (двох рідких й однієї парової), згідно з правилом фаз, має один ступінь вільності. Звідси випливає, що кожній температурі суміші строго відповідає певний тиск, і кожний компонент суміші поводиться незалежно від іншого. Відповідно парціальний тиск кожного компонента не залежить від його вмісту в суміші і дорівнює тиску пари чистого компонента за тієї самої температури.
На діаграмі р – х (рис. 3.6) лінії парціальних тисків і загального тиску є прямими, паралельними осями абсцис, причому рА = РА і рВ = РВ, а загальний тиск Р пари над сумішшю дорівнює сумі тиску пари чистих компонентів:
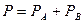 . (3.5)
. (3.5)
На рис. 3.6 наведено також фазові діаграми t – х – у і у – х для бінарних сумішей із взаємно нерозчинних компонентів. Пунктирними лініями на рисунку показано незначну розчинність компонентів суміші один в одному, яка фактично завжди є у рідин, які вважаються повністю взаємно нерозчинними, які зазвичай не враховується (рівняння (3. 5)).
На діаграмі t – х – у величини tA і tB – температури кипіння чистих компонентів А і В, 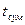 – температура кипіння суміші. З діаграми видно, що температура кипіння таких сумішей при Р = const є сталою і не залежить від складу розчину (пряма аb єпаралельною до осі абсцис). Температура кипіння завжди нижча за температуру кипіння чистих компонентів, що становлять суміш. Ця властивість використовується в техніці для розділення нерозчинних у воді рідин за допомогою перегонки з водяною парою.
– температура кипіння суміші. З діаграми видно, що температура кипіння таких сумішей при Р = const є сталою і не залежить від складу розчину (пряма аb єпаралельною до осі абсцис). Температура кипіння завжди нижча за температуру кипіння чистих компонентів, що становлять суміш. Ця властивість використовується в техніці для розділення нерозчинних у воді рідин за допомогою перегонки з водяною парою.
Для взаємно нерозчинних рідин склад пари над киплячою сумішшю є сталим і не залежить від співвідношення компонентів у розчині. Відповідно до закону Дальтона маємо:
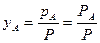 ,
, 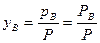 .
.
Отже, в цьому разі
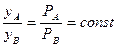 . (3.6)
. (3.6)
Співвідношення між кількостями GA і GB компонентів у парі таке:
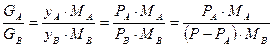 , (3.7)
, (3.7)
де Ма і Mb – молекулярна маса компонентів А і В.
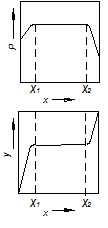
|
| Рис. 3.7. Діаграми р – х і у – х для сумішей рідин, обмежено розчинних одна в одній |
Суміші рідин, обмежено розчинних
одна в одній
Для таких сумішей у разі додавання однієї рідини до іншої (наприклад, фенолу до води) вони спочатку повністю розчиняються одна в одній, потім в певних межах концентрацій утворюють два шари (розчину) і поводяться подібно до взаємно нерозчинних рідин: змінюються їх кількості, але залишаються постійними склади рідких фаз. Тільки за підвищення концентрації розчиненої речовини (фенолу у воді) понад деяку межу один з шарів зникає і залишається одна рідка фаза – розчин води у фенолі. Отже, рідини знову стають необмежено взаємно розчинними.
Ці суміші найчастіше розділяють екстрагуванням, і їх властивості докладніше розглянуто нижче.
На рис. 3.7 наведено діаграми р – х і у – х для сумішей з обмеженою взаємною розчинністю. З графіків видно, що в межах концентрацій х1 і х2 такі суміші ідентичні до сумішей рідин, нерозчинних одна в одній.
3.3. Проста перегонка
Фракційна перегонка.Перегонку здійснюють поступовим випаровуванням рідини, що знаходиться в перегонному кубі. Пара, що утворюється, відводиться і конденсується. Процес здійснюють періодично або безперервно.
Якщо проста перегонка здійснюється періодично, то під час відгонки НК вміст його в кубовій рідині зменшується. Водночас змінюється в часі склад дистиляту, який збіднюється НК у міру перебігу процесу. Тому відбирають декілька фракцій дистиляту, що мають різний склад. Просту перегонку, що здійснюється з отриманням кінцевого продукту різного складу, називають фракційною або дробовою перегонкою.
У періодично діючій установці для фракційної перегонки (рис. 3.8) початкову суміш завантажують у перегонний куб 1, забезпечений змійовиком для обігріву, і доводять до кипіння. Пару відводять в конденсатор-холодильник 2. Фракції дистиляту надходять через оглядовий ліхтар 3 в окремі збірники 4 – 6. Після закінчення операції залишок зливають з куба, після чого в нього знов завантажують суміш, що розділяється.
Для складання матеріального балансу простої перегонки приймемо, що в кубі в деякий момент часу t міститься L кг перегонної суміші, що має поточну концентрацію х (за низькокиплячим компонентом). Маса НК в рідині у цей момент дорівнює Lx.
Нехай за нескінченно малий проміжок часу dx випарується dL кг суміші і концентрація рідини в кубі зменшиться на величину dx. При цьому утворюються dL кг пари, що є рівноважною з рідиною і має концентрацію у*; кількість НК в парі дорінюватиме dLy*. Відповідно залишок рідини в кубі становитиме (L – dL), кг, а її концентрація дорівнюватиме (х – dx). Тоді матеріальний баланс за НК виразиться рівнянням
Lx = (L – dL)×(x – dx) + dLy*.
Розкриваючи дужки і нехтуючи добутком dL×dx як нескінченно малою величиною другого порядку, після розділення змінних отримаємо
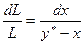
Рис. 3.8. Схема установки для фракційної перегонки: 1 – перегонний куб;
2 – конденсатор-холодильник; 3 – оглядовий ліхтар;
4–6 – збірники фракцій дистиляту
Це диференціальне рівняння необхідно проінтегрувати в межах зміни маси рідини в кубі від початкової L = F до кінцевої L = W (де F – маса початкової суміші, або живлення, і W – маса залишку) і відповідного зменшення її концентрації від 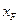 до
до 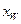 за всю операцію перегонки:
за всю операцію перегонки:
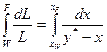 .
.
У результаті інтегрування отримаємо
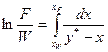 . (3.8)
. (3.8)
Вид функції у* = f(х) визначається формою кривої рівноваги і не може бути встановлений аналітично для кожного конкретного випадку перегонки. Тому інтегрування правої частини рівняння (3.8) виконують графічно – побудовою залежності 1/(у* – х) від х.
Для багатьох значень х у межах від до знаходять з діаграми у – х рівноважні їм значення у* і за розміром площі під кривою, обмеженою абсцисами і , визначають (з урахуванням масштабів діаграми) значення шуканого інтеграла. За рівнянням (3.8), знаючи маси F завантаженої в куб суміші і її склад , а також заданий склад залишку , знаходять масу залишку W. Маса перегнаної рідини становить F – W.
Середній склад (хP)сер одержуваного дистиляту розраховують з рівняння матеріального балансу за низькокиплячим компонентом:
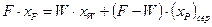 ,
,
звідки
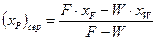 . (3.9)
. (3.9)
Розрахунок простої перегонки зазвичай має на меті визначення маси рідини, яку необхідно перегнати для того, щоб отримати в кубі залишок заданого складу і дистилят необхідного середнього складу.
Просту перегонку здійснюють за атмосферного тиску або під вакуумом, приєднуючи збірники дистиляту до джерела вакууму. Застосування вакууму дає можливість розділяти термічно малостійкі суміші і, внаслідок пониження температури кипіння розчину, використовувати для обігрівання куба пару нижчих параметрів.
Проста перегонка з дефлегмацією. Для підвищення ступеня розділення суміші перегонку здійснюють дефлегмацією, додатково збагачуючи дистилят. Пара з перегонного куба 1 (рис. 3.9) надходить в дефлегматор 2, де вона частково конденсується. З пари конденсується переважно ВК, і одержана рідина (флегма) зливається в куб. Пара, збагачена НК, прямує в конденсатор-холодильник 3, де повністю конденсується. Дистилят збирається в збірниках 4–6. Закінчення операції контролюють за температурою кипіння рідини в кубі, яка повинна відповідати заданому складу залишку. Останній видаляється з куба через штуцер 7.
Перегонка в струмені носія. Зниження температури кипіння суміші, що розділяється, можна досягти не тільки під час перегонки під вакуумом, а й внесенням в цю суміш додаткового компонента-носія (водяної пари або інертного газу).
Перегонка з водяною парою. Якщо компоненти початкової суміші є нерозчинними у воді, то її використовують як додатковий компонент, який вводять в куб зазвичай у вигляді гострої пари.
Під час перегонки висококиплячих речовин, нерозчинних у воді, з водяною парою, температура кипіння суміші повинна бути нижчою від температури кипіння води за даного тиску. Отже, за тиску, що дорівнює 1 атм, температура перегонки буде нижчою за 100 °С.
Цим способом зазвичай розділяють (або очищують від домішок) суміші речовин, що киплять за температур, вищих від 100°С, що зумовлює необхідність подавання води в куб у вигляді гострої перегрітої пари (рис. 3.10).
Початкову суміш завантажують в куб 1, що обігрівається глухою парою через сорочку. Всередину куба через барботер 2 подають гостру пару. Пара, що утворилася під час випаровування суміші, надходить в конденсатор-холодильник 3. Конденсат, що утворюється тут, через оглядовий ліхтар 4 надходить на розділення в сепаратор 5. Знизу сепаратора через гідравлічний затвор видаляється, наприклад, вода, а згори – відігнаний, не розчинний у воді легший компонент, який зливається в збірник 6.
Практично перегонка з водяною парою здійснюється в нерівноважних умовах. У цьому процесі гостра пара має подвійне приначення – теплоносія і агента, що знижує температуру кипіння. Тому пару треба подавати в кількості, більшій від її теоретичної витрати на процес власне відгонки виділеної речовини.
Процес можна розглядати як перегонку в потоці носія – водяної пари, яка може проводитися періодично (як показано на рис. 3.10) або безперервно. Загальна витрата тепла під час перегонки з водяною парою більша, ніж за простої перегонки, на кількість тепла, що видаляється з носієм (водяною парою).
Перегонка з інертним газом. Під час перегонки сумішей замість водяної пари іноді використовують інертні гази, наприклад, азот, двоокис вуглецю та ін. Перегонка в потоці інертного газу, що не конденсується, дає змогу більше понизити температуру випаровування суміші, що розділяється, ніж під час перегонки в потоці водяної пари, де це зниження обмежене температурою його конденсації. Водночас, присутність інертного газу в парі, що піднімається з куба, призводить до різкого зменшення коефіцієнта тепловіддачі в конденсаторі-холодильнику і, відповідно, до значного зростання поверхні теплообміну. Крім того, конденсація парогазових сумішей часто супроводжується туманоутворенням. Це вельми ускладнює розділення сумішей і спричиняє помітне винесення кінцевого продукту з інертним газом.
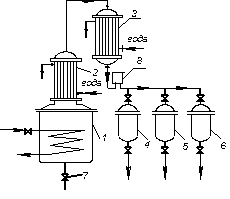
| 
|
| Рис. 3.9. Схема установки для простої перегонки з дефлегмацією: 1 – перегонний куб; 2 – дефлегматор; 3 – конденсатор-холодильник; 4–6 – збірники; 7 – штуцер для видалення залишку; 8 – оглядовий ліхтар | Рис. 3.10. Схема установки для перегонки з водяною парою: 1 – куб з паровою сорочкою; 2 – барботер для гострої пари; 3 – конденсатор-холодильник; 4 – оглядовий ліхтар; 5 – сепаратор; 6 – збірник продукту |
Теоретичну витрату водяної пари або інертного газу можна знайти із співвідношення (3.7). Зазвичай пара повністю не насичується компонентом, що вивільняється. Тому, враховуючи ступінь насичення пари (газу), який визначається коефіцієнтом насичення j = 0,5–0,9, витрату носія розраховують за рівнянням
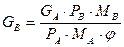 , (3.10)
, (3.10)
де всі позначення величин ті самі, що й в рівнянні (3.7).
У рівнянні (3.10) не враховується витрата носія, який є необхідним для нагрівання суміші до температури перегонки, випаровування суміші і компенсації втрат тепла у навколишнє середовище. Щоб уникнути надмірно великої витрати носія і розбавлення ним пари, тепло для вказаних вище випадків підводять в куб глухою парою або іншим теплоносієм.
Температуру перегонки з інертним газом або водяною парою можна визначити, користуючись графіком залежності тиску пари чистих компонентів від температури (рис. 3.11). Наносячи на графік криву тиску пари РВ = f (t) носія (наприклад, водяної пари) «обернено», тобто не від нуля, а від тиску, що дорівнює атмосферному, вниз, знаходять точку а перетину кривої тиску насиченої пари носія і відповідної рідини. Абсциса точки перетину вказує шукану температуру перегонки tn.
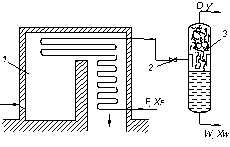
Рівноважна перегонка. Цей вид перегонки застосовується, наприклад, у нафтопереробній і нафтохімічній промисловості переважно для попереднього (перед ректифікацією) розділення складних сумішей, що містять леткі компоненти.
Початкова суміш нагрівається і одноразово випаровується в трубчастій печі 1, показаній на рис. 3.12 (D – кількість пари, решта позначень – попередні). До кінця процесу пара тривало контактує з рідиною, що не випарувалася, і стає в рівновазі з нею. Після досягнення кінцевої температури суміш пари і рідини через дросельний вентиль 2 прямує в сепаратор 3, де пара, що утворилася, відділяється від рідини, що не випарувалася. В такому процесі не досягається чіткого розділення суміші. Тому одержані продукти зазвичай піддають подальшій ректифікації.
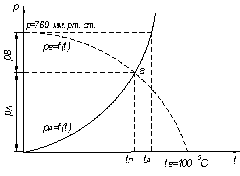
| 
|
| Рис. 3.11. До визначення температури перегонки з водяною парою | Рис. 3.12. Схема установки для рівноважної перегонки: 1 – трубчаста піч; 2 – дросельний вентиль; 3 – сепаратор |
Молекулярна перегонка. Для вилучення дорогих компонентів з сумішей, де вони містяться в невеликих кількостях, або для ретельного очищення термічно нестійких сумішей від домішок використовують молекулярну перегонку, що здійснюється в умовах глибокого вакууму. Цей процес відрізняється специфічними особливостями, потребує застосування спеціальної апаратури і буде розглянутий нижче.
3.4. Ректифікація
Принцип ректифікації.Як вже наголошувалося, високий ступінь розділення однорідних рідких сумішей на компоненти може бути досягнутий ректифікацією. Сутність процесів, з яких складається ректифікація, і одержувані при цьому результати можна прослідкувати за допомогою t – х – у діаграми
(рис. 3.13).
Нагріваючи початкову суміш складу х1 до температури кипіння, отримуємо пару, що знаходиться в рівновазі з рідиною, (точка b). Відбір і конденсація цієї пари дають можливість отримати рідину складу х2, збагачену НК (x2> x1). Нагріваючи цю рідину до температури кипіння t2, отримаємо пару (точка d), конденсація якої дає рідину з ще більшим вмістом НК, що має склад x3, і т.д. Проводячи послідовно багато процесів випаровування рідини і конденсації пари, в результаті можна отримати рідину (дистилят), що є практично чистим НК.
Подібно, виходячи з парової фази, що відповідає складу рідини х4, здійсненням декількох послідовних процесів конденсації і випаровування можна отримати рідину (залишок), що складається майже цілком з ВК.
У найпростішому вигляді процес багатократного випаровування можна здійснити в багатоступінчатій установці, в першому ступені якої випаровується початкова суміш. На другий ступінь надходить на випаровування рідина, що залишилася після виділення пари в першому ступені, в третьому ступені випаровується рідина, що надійшла з другого ступеня (після відбору з останньої пари), і т.д. Подібно можна організувати процес багатократної конденсації, під час якого на кожний наступний ступінь надходить для конденсації пара, що залишилася після видалення від неї рідини (конденсату) в попередньому ступені.
За достатньо великої кількості ступенів так можна одержати рідку або парову фазу з достатньо високою концентрацією компонента, яким вона збагатилася. Проте вихід цієї фази буде малий відносно її кількості в початковій суміші. Крім того, описані установки відрізняються громіздкістю і великими втратами тепла у навколишнє середовище.
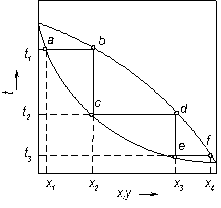
Рис 3.13. Зображення процесу розділення бінарної суміші ректифікацією
на діаграмі t – x – у
Більш економічного, повного і чіткого розділення сумішей на компоненти можна досягти в процесах ректифікації, що здійснюються зазвичай в компактніших апаратах – ректифікаційних колонах.
Процеси ректифікації здійснюються періодично або безперервно за різного тиску: за атмосферного тиску, під вакуумом (для розділення сумішей висококиплячих речовин), а також під тиском, більшим від атмосферного (для розділення сумішей, що є газоподібними за нормальних температур).
3.4.1. Схеми установок ректифікацій для розділення бінарних сумішей
Безперервно діючі установки. Розглянемо, як реалізуються вказані вище умови в ректифікаційних колонах безперервної дії (рис. 3.14), які здебільшого використовують у промисловості.
Ректифікаційна колона 1 має циліндричний корпус, усередині якого встановлено контактні пристрої у вигляді тарілок або насадки (конструкції тарілок і типи насадок, що використовуються, не відрізняються від описаних у другому розділі). Від низу до верху колоною рухається пара, що надходить у нижню частину апарата з кип’ятильника 2, який знаходиться ззовні колони, тобто є виносним (як показано на рис. 3.14), або розміщується безпосередньо під колоною. Отже, за допомогою кип’ятильника створюється висхідний потік пари.
Пара проходить крізь шар рідини на нижній тарілці, яку вважатимемо першою, починаючи нумерацію тарілок умовно від низу до верху.
Нехай концентрація рідини на першій тарілці дорівнює х1 (за низько киплячим компонентом), а її температура t1. У результаті взаємодії між рідиною і парою, що має вищу температуру, рідина частково випаровується, причому в пару переходить переважно НК. Тому на наступну (другу) тарілку надходить пара із вмістом НК у1 > х1.
Випаровування рідини на тарілці відбувається за рахунок тепла конденсації пари. З пари конденсується і переходить в рідину переважно ВК, вміст якого в парі, що надходить на тарілку, вищий від рівноважного зі складом рідини на тарілці. За рівності теплот випаровування компонентів бінарної суміші для випаровування 1 моль НК необхідно сконденсувати 1 моль ВК, тобто фази на тарілці обмінюються еквімолекулярними кількостями компонентів.
На другій тарілці рідина має склад х2, містить більше НК, ніж на першій (х2 > х1), і відповідно кипить за нижчої температури (t2 < t1). Контактуючи з нею, пара зі складом у1 частково конденсується, збагачуючись НК і видаляється на вищерозташовану тарілку, маючи склад у2 > х2, і т. д.
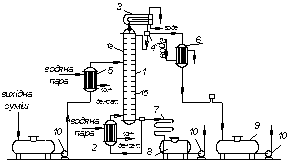
Рис. 3.14. Схема безперервно діючої установки ректифікації:
1 – ректифікаційна колона (1а – зміцнювальна частина; 1б – вичерпна частина);
2 – кип’ятильник; 3 – дефлегматор; 4 – дільник флегми; 5 – підігрівач початкової суміші; 6 – холодильник дистиляту або холодильник-конденсатор; 7 – холодильник залишку (або нижнього продукту); 8, 9 – збірники; 10 – насоси
Отже, пара, що є на виході з кип’ятильника майже чистим ВК, піднімаючись вгору, все більше збагачується низькокиплячим компонентом і залишає верхню тарілку колони у вигляді майже чистого НК, який практично повністю переходить в парову фазу на шляху пари від кип’ятильника до верху колони.
Пара конденсується в дефлегматорі 3, щоохолоджується водою, і одержувана рідина розділяється в дільнику 4 на дистилят і флегму, яка прямує на верхню тарілку колони. Отже, за допомогою дефлегматора в колоні створюється низхідний потік рідини.
Рідина, що надходить на зрошування колони (флегма), є майже чистим НК. Проте, стікаючи колоною і взаємодіючи з парою, рідина збагачується ВК, що конденсується з пари.
Коли рідина досягає нижньої тарілки, вона стає практично чистим ВК і надходить в кип’ятильник, що обігрівається глухою парою або іншим теплоносієм.
На деякій відстані від верху колони до рідини з дефлегматора приєднується початкова суміш, яка надходить на так звану живлячу тарілку колони. Для того, щоб зменшити теплове навантаження кип’ятильника, початкову суміш зазвичай заздалегідь нагрівають в підігрівачі 5 до температури кипіння рідини на живлячій тарілці.
Живляча тарілка умовно розділяє колону на дві частини різного призначення. У верхній частині 1а (від живлячої до верхньої тарілки) повинно бути забезпечене якомога більше зміцнення пари, тобто збагачення її НК з тим, щоб у дефлегматор прямувала пара, наближена за складом до чистого НК. Тому ця частина колони називається зміцнювальною. У нижній частині 16 (від живлячої до нижньої тарілки)необхідно максимально видалити з рідини НК, тобто вичерпати рідину для того, щоб в кип’ятильник стікала рідина, наближена за складом до чистого ВК. Ця частина колони називається вичерпною.
У дефлегматорі 3 може бути сконденсована або вся пара, що надходить з колони, або тільки частина її, відповідно до кількості, флегми, що повертається в колону. В першому випадку частина конденсату, що залишається після відокремлення флегми, є дистилятом (ректифікат), або верхнім продуктом, який після охолодження в холодильнику 6 прямує в збірник дистиляту 9. У другому випадку несконденсована в дефлегматорі пара водночас конденсується і охолоджується в холодильнику 6, якийза такого варіанта роботи є конденсатором-холодильником дистиляту.
Рідина, що виходить з низу колони (наближена за складом до ВК), також поділяється на дві частини. Одна частина, як вказано вище, прямує в кип’ятильник, а інша – залишок (нижній продукт) після охолоджування водою в холодильнику 7 прямує в збірник 8.
На рис. 3.14 наведено тільки принципову схему безперервно діючої установки ректифікації. Такі установки оснащуються необхідними контрольно-вимірювальними і регулювальними приладами, що дають можливість автоматизувати їх роботу і проводити процес за допомогою програмного керування в оптимальних умовах. Автоматичним регулюванням зводяться до мінімуму коливання кількості, складу і температури початкової суміші, а також тиску, витрати гріючої пари і охолоджувальної води.
Періодично діючі установки.Увиробництвах невеликого масштабу використовують ректифікаційні установки періодичної дії (рис. 3.15).
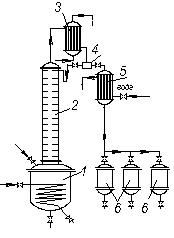
Рис. 3.15. Схема періодично діючої установки ректифікації:
1 – куб; 2 – ректифікаційна колона; 3 – дефлегматор; 4 – дільник флегми;
5 – холодильник; 6 – збірники дистиляту
Початкову суміш завантажують у куб 1, оснащений нагрівальним пристроєм. Суміш підігрівають до кипіння, і її пара надходить під нижню тарілку ректифікаційної колони 2. Піднімаючись по колоні, пара збагачується НК, яким збіднюється стікаюча донизу флегма, що надходить з дефлегматора 3 на верхню тарілку колони. Пара з колони прямує в дефлегматор 3, де вона повністю або частково конденсується. У разі повної конденсації рідина розділяється в дільнику 4 на флегму і дистилят. Кінцевий продукт (дистилят) охолоджують в холодильнику 5 іскеровують у збірники 6.
Після того, як досягнуто заданого складу залишку в кубі (про це роблять висновок за температурою кипіння рідини в ньому), залишок зливають, завантажують куб початковою сумішшю і операцію повторюють.
Порівнюючи періодично діючу колону (рис. 3.15) з ректифікаційною колоною безперервної дії (рис. 3.14), можна зауважити, що перша працює подібно до верхньої частини неперервно діючої колони як колона для зміцнення пари, а куб має роль вичерпної частини.
3.4.2. Матеріальний і тепловий баланси безперервної ректифікації бінарних сумішей
3.4.2.1. Основні положення
Мольні теплоти випаровування компонентів бінарної рідкої суміші зазвичай наближені за величинами, тоді як питомі теплоти випаровування (на 1 кг компонента) істотно відрізняються одна від однієї. Тому кількості і склади фаз під час аналізу і розрахунку ректифікації зручно подавати в мольних величинах. Відповідно до цього кількості рідин і їх пари подамо в кіломолях, а їх склади – у мольних частках НК.
Зробимо припущення, які мало спотворюють фактичні умови перебігу процесу, але значно спрощують розрахунок:
1. Суміш, що розділяється, відповідає правилу Трутона, згідно з яким відношення мольної теплоти випаровування або конденсації r до абсолютної температури кипіння Т для всіх рідин є приблизно величиною сталою. Для суміші, що містить п компонентів:
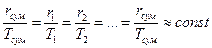
або за
Tсyм = T1 = T2 = … = Tn; rсyм = r1 = r2 = … .= rn.
2. Склад пари yD, що видаляється з колони в дефлегматор, дорівнює складу дистиляту хР. При цьому припускається, що зміцнювальною дією дефлегматора під час конденсації в ньому пари можна знехтувати і прийняти yD = уP = хР, де уР – склад дистиляту в паровій фазі.
3. Склад пари yW, що піднімається з кип’ятильника в колону, дорівнює складу рідини xW, що стікає в кип’ятильник з нижньої частини колони. Приймаючи yW = xW, нехтують вичерпною дією кип’ятильника, тобто зміною складу фаз під час випаровування в ньому рідини.
4. Теплоти змішування компонентів суміші, що розділяється, дорівнюють нулю.
З пп. 1 і 4, випливає, що під час конденсації 1 кмоль ВК в колоні випаровується 1 кмоль НК, тобто кількість пари (в кіломолях), що піднімається колоною, є величиною сталою. Крім того, в розрахунках виходять з того, що матеріальні і теплові втрати відсутні і що суміш, що підлягає розділенню, надходить в колону нагрітою до температури кипіння на живлячій тарілці.
3.4.2.2. Матеріальний баланс ректифікаційної колони
Припустимо, згідно до схеми на рис. 3.16, в колону надходить F кмоль вихідної суміші, склад якої xF мол. часток НК. Згори з колони видаляється G кмоль пари, створюючої після конденсації флегму і дистилят. Кількість одержуваного дистиляту Р кмоль, його склад хР мол. часток НК. На зрошування колони повертається флегма в кількості Ф кмоль, причому її склад дорівнює складу дистиляту (хФ = хР мол. часток). Знизу з колони видаляється W кмоль залишку складу xW мол. часток НК.
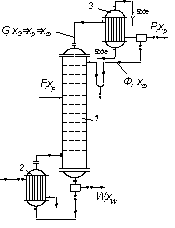
Рис. 3.16. До складання матеріального балансу ректифікаційної колони:
1 – колона; 2 – куб; 3 – дефлегматор
Тоді рівняння матеріального балансу колони буде:
F + Ф = G + W.
Оскільки G = Р + Ф, то
F = P + W (3.11)
Відповідно матеріальний баланс за НК:
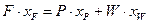 (3.12)
(3.12)
Рівняння робочих ліній.Для отримання рівнянь робочих ліній використовуємо загальне для всіх масообмінних процесів рівняння (3.11а), де виражаємо стосовно ректифікації концентрації, що в нього входять, у мольних частках:
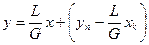 , (3.11а)
, (3.11а)
де L та G – витрати рідкої і парової фаз; 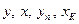 – відповідно поточні концентрації парової та рідкої фаз і їх концентрації в верхній частині колони.
– відповідно поточні концентрації парової та рідкої фаз і їх концентрації в верхній частині колони.
Рівняння (3.11а) можна представити також в іншому вигляді, якщо скористатися матеріальним балансом за розподілюваним компонентом:
Gyп + Lхп = Gyк+ Lx, (3.116)
звідки випливає, що
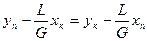 . (3.11в)
. (3.11в)
Після підстановки в рівняння (3.11а) отримаємо:
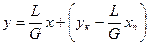 . (3.11г)
. (3.11г)
Зміцнювальна частина колони. Кількість рідини (флегми), що стікає цією частиною колони
L = Ф= Р·R. (3.12а)
Тут R = Ф/Р – флегмове число, що є відношенням кількості флегми до кількості дистиляту. Кількість пари, що піднімається колоною
G= P + Ф = P + P·R = P(R +1). (3.13)
Для верхнього кінця зміцнювальної частини колони склад пари yG = уP і, згідно з прийнятим вище припущенням, уР = хР. Отже, в цьому випадку 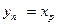 .
.
У тому самому перетині колони склад рідини (флегми), що надходить з дефлегматора, хФ = хР, тобто хК = хР. Підставляючизначення L, G 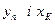 в рівняння (3.11а), отримаємо
в рівняння (3.11а), отримаємо
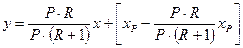 ,
,
звідки
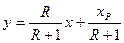 . (3.14)
. (3.14)
Залежність (3.14) є рівнянням робочої лінії зміцнювальної частини колони. У цьому рівнянні 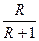 = tg а = А – тангенс кута нахилу робочої лінії до осі абсцис, а
= tg а = А – тангенс кута нахилу робочої лінії до осі абсцис, а 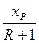 = В – відрізок, що відтинається робочою лінією на осі ординат діаграми у – х (рис. 3.17).
= В – відрізок, що відтинається робочою лінією на осі ординат діаграми у – х (рис. 3.17).
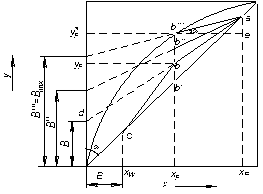
Рис. 3.17. Побудова робочих ліній ректифікаційної колони на у – х діаграмі
Вичерпна частина колони. Кількість зрошувальної рідини L/ в цій частині колони більше від кількості флегми Ф, що стікає зміцнювальною частиною на кількість початкової суміші, що надходить на живлячу тарілку. Якщо позначити кількість живлення, що припадає на 1 кмоль дистиляту через f = F/P, то F = P·f і кількість рідини, що стікає вичерпною частиною колони, становитиме
L/ = Ф + F = P·R + P·f = P (R +f).
Кількість пари, що проходить через нижню частину колони, дорівнює кількості пари, що підіймається по верхній (зміцнювальної) її частині. Отже
G/ = G = P×(R+1).
Для низу колони склад рідини (залишку), що видаляється х/k, = xW і, згідно з припущенням, склад пари, що надходить сюди з кип’ятильника у/n = yW = xW. Підставляючи значення L/ , G’, х/k і у/n у рівняння (3.11а), отримаємо
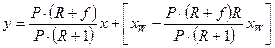 .
.
Після приведення до загального знаменника і скорочення подібних членів знаходимо:
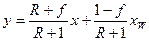 . (3.15)
. (3.15)
Залежність (3.15) є рівнянням робочої лінії вичерпної частини колони. У цьому рівнянні 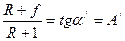 – тангенс кута нахилу робочої лінії до осі ординат, а
– тангенс кута нахилу робочої лінії до осі ординат, а 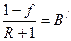 – відрізок, що відтинає робоча лінія на осі абсцис (рис 3.17).
– відрізок, що відтинає робоча лінія на осі абсцис (рис 3.17).
Помножуючи чисельник і знаменник виразів для A/ і А (для зміцнювальної частини колони) на кількість дистиляту Р, можна зауважити, що вони є відношенням кількостей рідкої і парової фаз, або питомою витратою рідини, що зрошує цю частину колони.
Побудова робочих ліній на діаграмі у – х. Для побудови робочих ліній відкладають на осі абсцис діаграми (рис. 3.17) задані склади рідин xW , хF і хP. Враховуючи прийняті припущення про рівність складів пари і рідини на кінцях колони, з точки хР проводять вертикаль до перетину з діагоналлю діаграми в точці а з координатами уР = хР.
Величину R вважаємо відомою. Відкладаючи на осі ординат відрізок 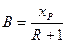 (рівняння (3.14)), сполучають прямий кінець відрізка (точку d) з точкою а. З точки, що відповідає заданому складу xF, проводять вертикаль до перетину з лінією ad в точці b. Пряма ab – робоча лінія зміцнювальної частини колони. Згідно з припущенням yW = xW, з точки, що відповідає складу xW, проводять вертикаль до перетину з діагоналлю діаграми і одержують точку с – кінцеву точку робочої лінії вичерпної частини колони. Сполучають точку с прямою з точкою b, що належить одночасно робочим лініям зміцнювальної і вичерпної частин колони. Пряма bc є робочою лінією вичерпної частини колони.
(рівняння (3.14)), сполучають прямий кінець відрізка (точку d) з точкою а. З точки, що відповідає заданому складу xF, проводять вертикаль до перетину з лінією ad в точці b. Пряма ab – робоча лінія зміцнювальної частини колони. Згідно з припущенням yW = xW, з точки, що відповідає складу xW, проводять вертикаль до перетину з діагоналлю діаграми і одержують точку с – кінцеву точку робочої лінії вичерпної частини колони. Сполучають точку с прямою з точкою b, що належить одночасно робочим лініям зміцнювальної і вичерпної частин колони. Пряма bc є робочою лінією вичерпної частини колони.
Робочі лінії аb і bс, на відміну від робочих ліній процесу абсорбції, розміщаються під лінією рівноваги. Тоді, як вже наголошувалося, НК переходить в парову фазу, прагне до рівноваги з рідкою фазою, тобто десорбує з рідини.
3.4.2.3. Мінімальне і дійсне флегмове число
Розрахунок мінімального флегмового числа. За заданого складу дистиляту хР величина відрізка В (рис. 3.17), що відтинається робочою лінією зміцнювальної частини колони на осі ординат, залежить тільки від флегмового числа R, оскільки . Із зменшенням R відрізок В збільшується (В/ > В), і робоча лінія ніби повертається навколо точки а за годинниковою стрілкою, займаючи послідовно положення ab/, ab// і т.д. Проте величину R можна зменшувати тільки до деякої межі, що визначається рушійною силою процесу масоперенесення між рідкою і паровою фазами.
Рушійна сила, виражена в концентраціях парової фази, зображається на діаграмі у – х вертикальним відрізком між заданою точкою на робочій лінії і лінією рівноваги. Наприклад, при робочій лінії аb в точці введення живлення (xF) рушійна сила дорівнює 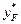 – yF і зображається відрізком b///b. Із зменшенням R точка b зміщується по вертикалі, що відповідає абсцисі точки, яка відповідає складу xF, і рушійна сила знижується доти, поки не перетвориться на нуль (точка b/// ). При цьому робоча лінія аb/// відтинає на осі ординат максимальний відрізок В/// = Bmax, якому за заданого хР відповідає мінімальне флегмове число Rmin:
– yF і зображається відрізком b///b. Із зменшенням R точка b зміщується по вертикалі, що відповідає абсцисі точки, яка відповідає складу xF, і рушійна сила знижується доти, поки не перетвориться на нуль (точка b/// ). При цьому робоча лінія аb/// відтинає на осі ординат максимальний відрізок В/// = Bmax, якому за заданого хР відповідає мінімальне флегмове число Rmin:
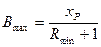 .
.
Зауважимо, що в деякій точці на вертикалі, що відповідає xF і розміщена вище від лінії рівноваги, робочі лінії перетнутися не можуть, оскільки в цьому випадку рушійна сила процесу мала б негативне значення, що суперечить фізичному змісту.
Із збільшенням R відрізки В зменшуються, і робоча лінія повертається навколо точки а проти годинникової стрілки. Вочевидь, нижнє граничне положення робочих ліній повинне відповідати збігу точки їх перетину з діагоналлю діаграми (точка b/). При цьому кут нахилу робочих ліній до осі абсцис дорівнює 45°, А = А’ = 1 і В = В’ = 0, що є можливим, як випливає з виразів для В і В’, тільки за нескінченно великого флегмового числа (R = ¥).
Дійсне (робоче) флегмове число Rд, за якого працює колона, повинно знаходитися в межах Rmln і R = ¥. Вихідною величиною для вибору дійсного флегмового числа є Rmin, значення якого можна знайти розрахунком.
Для визначення Rmin проведемо з точки b/// (рис. 3.17) горизонтальний відрізок b///е до перетину з ординатою точки а. Тангенс кута нахилу робочої лінії зміцнювальної частини колони за Rmin дорівнює відношенню катетів ае і b///е трикутника ab"’e, причому катет ае = уР – = хР – , а катет b///е = xP – xF. Отже,
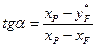 . (А)
. (А)
Разом з тим, згідно з рівнянням (3.14), за мінімального флегмового числа
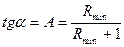 . (Б)
. (Б)
Зіставляючи вирази (А) і (Б), отримаємо
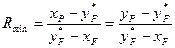 . (3.16)
. (3.16)
Розрахунок дійсного флегмового числа. Раціональний вибір дійсного флегмового числа є складною задачею. Це пояснюється тим, що флегмове число R визначає загалом розміри апарата і витрати теплоносіїв (гріючого агента в кип’ятильнику, охолоджувальної води в дефлегматорі). Отже, від величини R залежать капітальні витрати і експлуатаційні витрати на ректифікацію.
Експлуатаційні витрати, що визначаються витратою теплоносія, зростають прямо пропорційно до величини R (рис. 3.18, крива 1). Складнішою є залежність капітальних витрат від величини флегмового числа. Із збільшенням R зростає рушійна сила процесу і зменшується необхідна кількість теоретичних і відповідно дійсних ступенів. У результаті за деякого флегмового числа робочий об’єм колони стане мінімальним і, отже, мінімальною буде її вартість. Тому залежність капітальних витрат від флегмового числа має мінімум (к
Дата добавления: 2015-07-24; просмотров: 10943;