Періодична ректифікація бінарних сумішей
Процеси періодичної ректифікації можуть здійснюватись: 1) за постійного флегмового числа (R = const); 2) за постійного складу дистилята (хР = const).
За періодичної ректифікації вміст НК всуміші, яка кипить в кубі, зменшується в часі. Тому під час повернення сталої кількості флегми в колону, тобто у разі роботи за R = const, дистилят також поступово збіднюється НК. В результаті дистилят одержують у вигляді різних за складом фракцій, відбираних в окремі збірники (рис. 3.15).
Здійснення процесу з отриманням дистиляту сталого (початкового) складу хР=const можливе поступовим збільшенням в часі кількості флегми, що повертається або при роботі зі поступово зростаючим числом R.
Здійснення процесу у такий спосіб пов’язане з автоматичним (програмованим) регулюванням кількості флегми, що повертається в колону, або кількості пари, яка надходить з кип’ятильника, що ускладнює конструкцію установки.
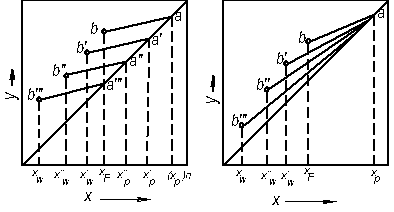
Як наголошувалося, періодично діючі колони працюють як колони для зміцнення пари. Тому залежність між робочими концентраціями фаз визначається для всієї колони однією робочою лінією відповідно до рівняння (3.14). Роль вичерпуючої частини виконує куб колони. Під час періодичної ректифікації за R = const концентрація НК в кубі поступово зменшується від xF (в початковий момент) до xW (в кінцевий момент), приймаючи в часі проміжні значення x/W i x//W і т.д. (рис. 3.21, а). За R = const (рис. 3.21, а) нахил робочої лінії, дорівнює 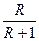 , не залежить від концентрації, і тому робоча лінія зміщується паралельно до свого первинного положення.
, не залежить від концентрації, і тому робоча лінія зміщується паралельно до свого первинного положення.
Проте за всіх положеннь робочої лінії кількість одиниць перенесення в колоні залишається незмінною. Тому змінюється в часі склад дистиляту; концентрація НК в ньому знижується, набуваючи послідовно значень (хР)n (за складу хF в кубі), х’р, х"р і т.д. аж до кінцевого значення 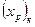 , що відповідаєзаданому складу залишку
, що відповідаєзаданому складу залишку 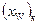 У результатіодержують дистилят, середній склад якого можна розраховувати за рівнянням:
У результатіодержують дистилят, середній склад якого можна розраховувати за рівнянням:
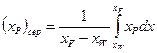 .
.
А б
Рис. 3.21. Зображення робочих ліній процесу періодичної ректифікації:
а – за R = const; б – за 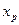 = const
= const
Інтеграл в правій частині цього рівняння визначають методом графічного інтегрування, користуючись графіком залежності між складом дистиляту і кубової рідини xP = f(х).
Під час періодичної ректифікації за хР = const для збереження сталого складу дистиляту необхідно поступово збільшувати флегмовое число. Його зростання відповідає збільшенню нахилу робочої лінії, тобто повороту її проти годинникової стрілки навколо точки а (рис 3.21, б). Робоча лінія займає послідовно положення ab, ab/, аb" і т. д., оскільки із збільшенням R відрізок, щовідтинається робочою лінією на осі ординат діаграми і дорівнює 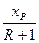 , за хР = const зменшується. Особливості розрахунку процесів періодичної ректифікації розглянуто нижче.
, за хР = const зменшується. Особливості розрахунку процесів періодичної ректифікації розглянуто нижче.
3.4.2.5. Ректифікація багатокомпонентних сумішей
У промисловості частіше розділяють не бінарні, а багатокомпонентні суміші, ректифікація яких є складнішим і менш вивченим процесом. На відміну від бінарних сумішей – систем, що мають тільки два ступені свободи, багатокомпонентна суміш є системою, кількість ступенів свободи якої дорівнює кількості компонентів, які її утворюють. Це ускладнює аналіз і розрахунок процесів ректифікації таких сумішей.
Якщо для бінарної суміші є відомими загальний тиск перегонки і мольна частка одного з компонентів в дистиляті, то, згідно з правилом фаз, цими умовами однозначно визначаються склад дистиляту і температура його конденсації. У разі ж розділення багатокомпонентної суміші, що складається з п компонентів і має п ступенів свободи, за заданих вказаних вище двох параметрах залишаються невідомими ще п – 2 ступеня свободи. Тому вміст решти компонентів в дистиляті можна знайти тільки підбором, враховуючи, що існує багато сумішей різного складу, які за заданого тиску мають однакову температуру кипіння.
Водночас під час розділення багатокомпонентних сумішей ускладнюється і апаратурне оформлення процесу ректифікації. Багатокомпонентну суміш не можна розділити в одній колоні, подібно до бінарної суміші. Загалом для ректифікації багатокомпонентної суміші має бути на одну колону менше, ніж кількість компонентів, на які розділюється суміш, тобто для розділення суміші з п компонентів необхідна п – 1 колона.
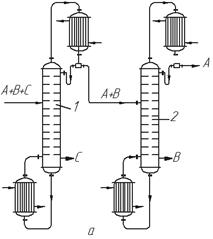
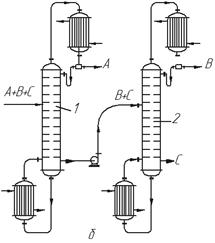
Розглянемо це на прикладі розділення суміші, що складається з трьох компонентів А, В і С, що не створюють азеотропів (рис. 3.22). За одним варіантом (рис. 3.22, а) в колоні 1 найменш леткий з компонентів (компонент С) виділяють у вигляді залишку. Інші два компоненти (В і А), що відводяться як дистилят, після конденсації надходять в колону 2, де розділяються на дистилят (компонент А) і залишок (компонент В менш леткий, ніж А). Економічніше подавати компоненти А + В в колону 2 у вигляді пари; при цьому в дефлегматорі першої колони конденсується тільки флегма, що є необхідною для зрошування колони.
За іншим варіантом (рис. 3.22б) в колоні 1 виділяють у вигляді дистиляту найлеткіший компонент А, а суміш двох інших (В + С) подають на розділення в колону 2. З цієї колони відносно леткіший компонент В одержують як дистилят, а компонент С є залишком.
Пристрої ректифікаційних апаратів
Для ректифікації використовують апарати різноманітних конструкцій, основні типи яких не відрізняються від відповідних типів абсорберів.
| 
|
| Рис. 3.22. Схеми установок для ректифікації трикомпонентних сумішей: а – компоненти А і В леткіші, ніж компонент С; б – компонент А леткіший, ніж компоненти В і С |
У ректифікаційних установках використовують переважно апарати двох типів: насадкові і тарілчасті ректифікаційні колони. Крім цього, для ректифікації під вакуумом використовують плівкові і роторні колони різних конструкцій.
Насадкові, барботажні, а також деякі плівкові колони за конструкцією внутрішніх пристроїв (тарілок, насадкових тіл та ін.) аналогічні абсорбційним колонам, розглянутим у розділі II. Проте, на відміну від абсорберів, ректифікаційні колони оснащені теплообмінними пристроями – кип’ятильником (кубом) і дефлегматором. Крім цього, для зменшення втрат тепла у довкілля ректифікаційні апарати покривають тепловою ізоляцією.
Кип’ятильник, або куб призначений для перетворення на пару частини рідини, що стікає з колони, і підведення пари в її нижню частину (під насадку або нижню тарілку). Поверхня нагрівання кип’ятильника виготовлена у вигляді змійовика (рис. 3.15) або кожухотрубного теплообмінника, вбудованого в нижню частину колони (рис. 3.23а). Зручнішими для ремонту і заміни є виносні кип’ятильники (рис. 3.14), які встановлюють нижче від колони для того, щоб забезпечити природну циркуляцію рідини.
У періодично діючих колонах куб є не тільки випарником, але й місткістю для початкової суміші. Тому об’єм куба повинен бути в 1,3 – 1,6 раза більшим ніж його разове завантаження (на одну операцію). Обігрівають кип’ятильники зазвичай насиченою водяною парою.
Дефлегматор, призначений для конденсації пари і подавання зрошування (флегми) в колону, є кожухотрубчастим теплообмінником, у міжтрубному просторі якого зазвичай конденсується пара, а трубами рухається охолоджувальний агент (вода). Проте питання про напрям пари і охолоджувального агента, що конденсується, всередину або зовні труб необхідно вирішувати у кожному конкретному випадку, враховуючи бажаність підвищення коефіцієнта теплоперенесення і зручність очищення поверхні теплообміну.
У разі часткової конденсації пари в дефлегматор його розміщують або поза колоною, (рис. 3.14) або безпосередньо над колоною (рис. 3.23а), щоб забезпечити компактність установки. Тоді конденсат (флегму) з нижньої частини дефлегматора подають безпосередньо через гідравлічний затвор на верх колони, оскільки в цьому разі відпадає необхідність у встановленні дільника флегми (рис. 3.15).
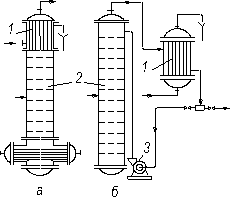
Рис. 3.23. Варіанти встановлення дефлегматора: а – на колоні;
б – нижче від верху колони; 1 – дефлегматори; 2 – колони; 3 – насос
У разі повної конденсації пари в дефлегматорі, його встановлюють вище від колони (рис. 3.14), безпосередньо на колоні (рис. 3.23а) або нижче від верху колони (рис. 3.23б) для того, щоб зменшити загальну висоту установки. В останньому випадку флегму з дефлегматора 1 подають в колону 2 насосом 3. Таке розміщення дефлегматора переважно використовують у разі встановлення ректифікаційних колон поза будівлями, що є економічнішим в умовах помірного клімату.
Барботажні колони. В процесах ректифікації переважно використовують ці апарати. Вони застосовні для великих виробників, широкого діапазону змін навантажень за парою і рідиною і можуть забезпечити чітке розділення сумішей. Вказанний вище (розділ II) недолік барботажних апаратів – відносно високий гідравлічний опір – в умовах ректифікації не має такого істотного значення, як в процесах абсорбції, де величина Dр пов’язана із значними витратами енергії на переміщення газу через апарат. Під час ректифікації підвищення гидравлічного опору приводить тільки до деякого збільшення тиску і відповідно до підвищення температури кипіння рідини в кип’ятильнику колони. Проте той самий недолік (значний гідравлічний опір) впливає на процеси ректифікації під вакуумом.
Насадкові колони. У цих колонах використовуються насадки різних типів (розділ II), але в промисловості це зазвичай кільця Рашига. Менший гідравлічний опір насадкових колон порівняно з барботажними є особливо важливим для ректифікації під вакуумом. Навіть за значного вакууму у верхній частині колони внаслідок великого гідравлічного опору розрідження в кип’ятильнику може бути недостатнім для необхідного зниження температури кипіння вихідної суміші. Для зменшення гідравлічного опору вакуумних колон в них застосовують насадки з якомога більшим вільним об’ємом.
У ректифікаційній колоні не ставиться вимога відводити тепло, як у абсорберах. Тому складність відведення тепла з насадкових колон є швидше перевагою, ніж їхнім недоліком в умовах процесу ректифікації.
Проте і під час ректифікації необхідно враховувати те, що рівномірний розподіл рідини по насадці в колонах великого діаметра є ускладненим. У зв’язку з цим діаметр промислових насадкових ректифікаційних колон зазвичай не перевищує 0,8–1 м.
Плівкові апарати.Ці апарати застосовують для ректифікації під вакуумом сумішей, що мають низьку термічну стійкість під час нагрівання (наприклад, різні мономери і полімери, а також інші продукти органічного синтезу).
У ректифікаційних апаратах плівкового типу досягають низького гідравлічного опору. Крім того, затримка рідини в одиниці об’єму працюючого апарату є малою.
До плівкових ректифікаційних апаратів належать колони з регулярною насадкою у вигляді пакетів вертикальних трубок з діаметром 6–20 мм (багатотрубчасті колони), а також пакетів плоскопаралельної або стільникової насадки з каналами різної форми, виготовленої з перфорованих металевих листів або металевої сітки.
Одну з поширених конструкцій роторно-плівкових колон показано на рис. 3.24. Вона складається з колони, або ректифікатора 1, забезпеченого зовнішнім обігріванням крізь парові оболонки 2 і ротором 3, роторного випарника 4 і конденсатора 5. Ротор, що є порожнистою трубою з лопатями, охолоджуваною зсе-
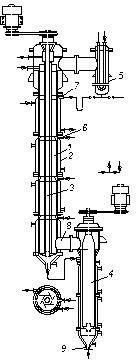
|
| Рис. 3.24. Схема роторно-плівкової ректифікаційної колони: 1 – колона; 2 – оболонка для обігрівання; 3 – ротор; 4 – роторний випарник; 5 – конденсатор-дефлегматор; 6 – штуцер для надходження вихідної суміші; 7 – штуцер для надходження флегми; 8 – штуцер для надходження пари; 9 – штуцер для виведення залишку |
редини водою, обертається усередині корпусу колони. Вихідна суміш надходить в колону через штуцер 6. Згори колона зрошується флегмою, що надходить з конденсатора 5 через штуцер 7. Пару подають в колону через штуцер 8 з випарника 4, забезпеченого неохолоджуваним ротором і аналогічного до плівкового випарного апарата. Підіймаючись у просторі між ротором 3 і корпусом колони 1, пара конденсується на зовнішній поверхні ротора. Плівка конденсату, що утворюється, відкидається під дією відцентрової сили по поверхні лопатей ротора до периферії. Потрапляючи на внутрішню поверхню, що обігрівається, рідина випаровується і пара, що утворюється, піднімається догори. Таким конденсаційно-випарним способом (за роботи колони в неадіабатичних умовах) досягають чіткого розділення суміші за нетривалого часу її перебування в апараті і незначного перепаду тиску за висотою колони, оскільки велика частина внутрішнього простору корпусу заповнена потоком пари. Роторні випарники типу випарника 4 можна використовувати як самостійні апарати для вакуумної дистиляції сумішей, чутливих до високих температур.
Недоліки роторних колон: обмеженість їх висоти і діаметра (через складність виготовлення і вимоги, що ставлять до міцності і жорсткості ротора), а також високі експлуатаційні витрати.
3.5. Розрахунок ректифікаційних апаратів
3.5.1. Безперервна ректифікація бінарних сумішей у насадкових колонах
Визначення фіктивної швидкості пари і діаметра колони. Швидкість пари, віднесену до всього перетину колони, вибирають, як вказувалось вище, залежно від запланованого гідродинамічного режиму роботи апарата. Зазвичай як початкову величину розраховують граничну швидкість пари, що відповідає точці «захлинання» і яку можна буде визначити за рівнянням:
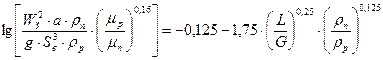 , (3.21)
, (3.21)
де Sв – вільний переріз насадки, 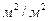 ; а – питома поверхня насадки,
; а – питома поверхня насадки, 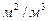 ; WЗ – швидкість пари в точці захлинання,
; WЗ – швидкість пари в точці захлинання, 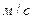 ; L та G – витрати рідини і пари, кг/с; rп та rр, mп та mр – густини (
; L та G – витрати рідини і пари, кг/с; rп та rр, mп та mр – густини ( 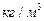 ) і в’язкості (
) і в’язкості ( 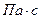 ) пари і рідини відповідно.
) пари і рідини відповідно.
Фіктивну швидкість пари знаходять, помноживши WЗ на коефіцієнт k, що є меншим від одиниці і залежить від вибраного гідродинамічного режиму. Наближено для режиму емульгування k = 0,85¸1, для режиму підвисання 0,45 £ k £ 0,85 і для плівкового режиму k £ 0,45.
У літературі [5]наводяться також інші розрахункові залежності для визначення фіктивної швидкості пари, що відповідає різним режимам роботи насадковних колон. Так, наприклад, фіктивну швидкість пари, що відповідає початку (точці) підвисання, рекомендується визначати за рівнянням:
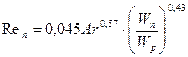 , (3.21а)
, (3.21а)
де 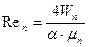 – критерій Рейнольдса в точці підвисання; Wп та Wр – масові швидкості пари і рідини;
– критерій Рейнольдса в точці підвисання; Wп та Wр – масові швидкості пари і рідини; 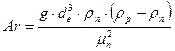 – критерій Архімеда, розрахований за еквівалентним діаметром de насадки.
– критерій Архімеда, розрахований за еквівалентним діаметром de насадки.
Діаметр колони розраховують за відомими значеннями витрати та швидкості пари. За великої відмінності витрат рідини в зміцнювальній і вичерпній частинах колони діаметр розраховують для кожної з цих частин, оскільки вичерпна частина звичайно має більший діаметр, чим зміцнювальна.
Визначення висоти насадки. Робоча висота насадки може бути визначена будь-яким із способів, описаних в розділі I для масообмінних апаратів з безперервним контактом фаз. Як наголошувалося, розрахунок на основі кількості одиниць перенесення можна виконати графоаналітичним або графічним методами, описаними в розділі I.
Загальну висоту насадки знаходять як суму висот насадок для зміцнювальної і вичерпної частин колони.
3.5.2. Безперервна ректифікація бінарних сумішей у тарілчастих колонах
Визначення фіктивної швидкості пари і діаметра колони. Максимально допустиму фіктивну швидкість пари для тарілчастих колон приймають нижчою від граничної, що відповідає точці захлинання тарілок (для колон, що працюють під атмосферним і надлишковим тиском), а також надмірно великим віднесенням рідини або перепаду тиску в колоні (для колон, що працюють під розрідженням). Максимально допустиму фіктивну швидкість пари визначають за формулою загального вигляду:
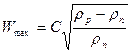 , (3.22)
, (3.22)
у якій коефіцієнт С залежить від типу тарілки, відстані між тарілками, навантажень за рідиною і парою і фізичними властивостями фаз.
Значення коефіцієнта С можна визначити за рівнянням:
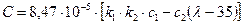 , (3.23)
, (3.23)
де величина k1 змінюється в межах 1¸1,4. Так, наприклад, для ковпачкових тарілок до k1 ~ 1, а для сітчастих тарілок k1 ~ 1,2 при Sв = 4–8 %.
Для тарілчастих колон, що працюють за атмосферного та надлишкового тиску, зазвичай k2= 1.
Величина с1 для ковпачкових тарілок (за відстані між ними 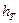 = 200-350 мм) визначається за формулою:
= 200-350 мм) визначається за формулою: 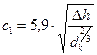 .
.
Для тарілок інших типів 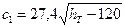 .
.
Величина l є функцією лінійної густини зрошування U .
При U < 10 м3/(м×год) – 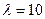 ; а при U > 65 м3/(м×год) –
; а при U > 65 м3/(м×год) – 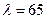 . Докладніші дані наводяться в спеціальній літературі [12].
. Докладніші дані наводяться в спеціальній літературі [12].
Визначення робочої висоти колони. Робочу висоту колони, що дорівнює відстані між крайніми тарілками, знаходять різними способами, вказаними в розділі I для масообмінних апаратів зі ступінчастим контактом фаз.
У розрахунковій практиці величину НТ найчастіше визначають через кількість пд реальних тарілок за формулою
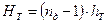 , (3.24)
, (3.24)
де hТ –відстань між тарілками.
Кількість реальних тарілок пд для кожної частини колони знаходять побудовою кінетичної кривої або розділенням загальної кількості одиниць перенесення для заданої частини колони на кількість одиниць перенесення, що припадає на одну тарілку. Одержану кількість тарілок підсумовують і одержують величину пд для колони.
Кількість одиниць перенесення на тарілку визначають для парової і рідкої фаз за правилом адитивності відповідно. Проте в цьому разі розрахунок є утрудненим через відсутність надійних залежностей для обчислення кількості одиниць перенесення на тарілку за кожною з фаз, тобто величин п/у і п/х. Тому, незважаючи на відмінність процесів абсорбції і ректифікації (як зазначалося, ректифікація відрізняється взаємним обміном компонентами між фазами в результаті одночасного перебігу процесів масо- і теплообміну), в першому наближенні величини п/у і п/х можна знайти за допомогою відповідних формул для тарілчастих абсорберів.
У розрахунковій практиці робочу висоту барботажних ректифікаційних колон іноді знаходять за кількістю теоретичних ступенів (тарілок). Розрахунок кількості цих ступенів, як було описане раніше, зводиться до побудови «сходинок» між лінією рівноваги і робочою лінією. За діаграмою у–х визначають кількість теоретичних ступенів для зміцнювальної (п/Т) і вичерпної (п//Т)частин колони. Розділюючи величину пТ = п/Т + n//Т насереднє значення ефективності (к.к.д.) колони Е, відповідно до виразу ( 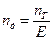 ) знаходять кількість дійсних тарілок
) знаходять кількість дійсних тарілок 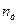 .
.
Такий розрахунок відрізняється простотою. Проте дотепер не отримані надійні рівняння для обчислення Е, що зумовлено складною залежністю ККД від багатьох чинників, а також від конструкції тарілок і взаємного напряму руху фаз на них, фізичних властивостей пари і рідини, швидкостей фаз, винесення рідини парою та ін.
Для розрахунку середньої загальної ефективності колони з ковпачковими або сітчастими (з переливними пристроями) тарілками рекомендується формула:
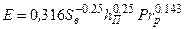 , (3.25)
, (3.25)
де SB – вільний перетин тарілки, м2/м2; hП – висота переливної планки на тарілці, м; Рrр = v/D – критерій Прандтля для рідини; n – кінематична в’язкість рідини, м2/год; D – коефіцієнт дифузії в рідині, м2/год.
Значення Е розраховують окремо для верхньої і нижньої частин колони.
3.5.3. Періодична ректифікація бінарних сумішей
Особливість розрахунку періодично діючих колон ректифікацій, що працюють за R = const, полягає в тому, що флегмовое число і кількості одиниць перенесення (або кількість теоретичних ступенів) визначають для початкового або кінцевого моменту процесу. Розрахунок виконують зазвичай графічним способом (див. розділ I) стосовно зміни концентрацій в межах від (хР)пдо xF (для початкового моменту) або від (хР)к до (хW) (для кінцевого моменту), причому відповідні дві граничні концентрації повиннібути заданими. Приймаючи довільно проміжні значення концентрацій х/Р, х//Р і т. д., проводять паралельно одна до однієї відповідні їм робочі лінії (рис. 3.21, а). Для кожного положення робочої лінії між нею і лінією рівноваги будують постійну кількість ступенів, визначену раніше (наприклад, для початкового моменту процесу) і так знаходять концентрації у кубовому залишку, що відповідають х/Р, х//Р і т.д.
Кількість кубової рідини GW, що залишається в кубі до кінця процесу, розраховують за рівнянням, ідентичним рівнянню (3.8) для простої перегонки, в якому у дорівнює хР – поточній концентрації дистиляту. При цьому інтеграл, що входить в рівняння, визначають графічно. За відомого GW для знаходження середнього складу дистиляту можна скористатися рівнянням (3.9).
Періодично діючі барботажні колони, що працюють за R=const, розраховують так само, будуючи кінетичні криві і визначаючи кількість реальних ступенів (тарілок) для початкового або кінцевого моменту процесу.
Розрахунок колон періодичної дії, що працюють з отриманням дистиляту сталого складу (хР =const), починають із знаходження флегмового числа і кількості одиниць перенесення для кінцевого моменту процесу. Потім, приймаючи довільно ряд менших значень R, будують для кожного з них робочу лінію і вписують між нею і лінією рівноваги кількість одиниць перенесення, визначену для кінця процесу, оскільки ця кількість в реальній колоні залишається незмінною в часі. Далі за діаграмою у–х знаходятьсклади кубової рідини, що відповідають прийнятим значенням R.
Середнє флегмовое число для всього процесу визначають методом графічної інтеграції за рівнянням, яке є подібним до виразу для розрахунку середнього складу дистиляту
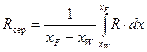 . (3.26)
. (3.26)
Для барботажних колон, що працюють періодично за = const, розрахунок починають з побудови кінетичної кривої і визначення кількості реальних ступенів (тарілок) для кінцевого моменту процесу. Потім будують кінетичні криві, задаючись меншими значеннями R, і за умови, що = const, знаходять склади рідини в кубі.
3.5.4. Ректифікація багатокомпонентних сумішей
Більшість сумішей, що розділяються в промисловості, містить більше двох компонентів. Проте у багатьох випадках суміші складаються переважно з двох компонентів, концентрації ж решти компонентів дуже малі порівняно із вмістом двох основних. Враховуючи це, подібні суміші можна розглядати як бінарні. Якщо ж таке допущення неможливе, завдання розрахунку багатокомпонентних сумішей значно ускладнюється. При цьому варто зазначити, що методи розрахунку ректифікації вказаних сумішей ще не є такими розробленими, як теорія і розрахунок розділення бінарних сумішей.
Під час розрахунку процесу ректифікації багатокомпонентної суміші необхідно визначити діаметр апарата, кількість одиниць перенесення (ступенів розділення), що необхідна для здійснення заданого розділення, флегмовое число, розподіл температур, потоків і концентрацій компонентів за висотою колони, теплові навантаження дефлегматора і кип’ятильника, а також встановити місце оптимального введення живлення в колону.
Всі існуючі методи розрахунку ректифікації багатокомпонентних сумішей можна розділити на точні і наближені. Зважаючи на недостатність даних про кінетику процесу масообміну під час багатокомпонентної ректифікації, розрахунок здійснюють зазвичай за кількістю теоретичних ступенів розділення, або теоретичних тарілок. Точні методи передбачають розрахунок від ступеня до ступеня, припускаючи, що суміш, що розділяється, є ідеальною. Наближені методи засновані на зведенні багатокомпонентної суміші до бінарної.
Точні методи полягають у розв’язанні різними способами системи рівнянь матеріального і теплового балансів, а також рівнянь термодинамічної рівноваги для кожного ступеня, причому розрахунок виконують послідовно від ступеня до ступеня. Розв’язання одержуваної системи нелінійних рівнянь алгебри високого порядку (кількість їх є пропорційною до кількості компонентів суміші, що розділяється, і кількості ступенів) є вельми трудомісткою задачею і тому вимагає застосування комп’ютерної техніки.
Такі задачі розв’язують методом послідовних наближень (ітерацій). У зв’язку з цим дуже важливе значення має вибір незалежних змінних і початкових даних для початкового наближення, а також критерію збіжності, що визначає напрям ітерацій.
Відомо два способи розрахунку від ступеня до ступеня, які відрізняються незалежними змінними: Люїса і Матісона (незалежні змінні – склади продуктів розділення) і Тіле і Геддеса (незалежні змінні – температури на кожному теоретичному ступені).
Решта величин, які повинні бути задані для розв’язання системи, наприклад, кількість і склад живлення, флегмове число і т. д., є вже залежними змінними. Тільки в цьому випадку забезпечується однозначний розв’язок системи нелінійних рівнянь алгебри, що описують процес розділення. Чим складніший процес ректифікації багатокомпонентної суміші (декілька введень живлення в колону і виводів продуктів розділення), тим більша кількість залежних змінних.
Різноманітність запропонованих методів розрахунку ректифікації (а також абсорбція) багатокомпонентних сумішей від ступеня до ступеня пов’язана переважно з другою умовою розв’язання рівнянь методом послідовних наближень – вибором початкових даних і критерію збіжності задачі. Проте, зважаючи на різноманіття сумішей і велику відмінність їх властивостей, жоден з методів розв’язання не можна вважати універсальним.
Точні методи розрахунку багатокомпонентної ректифікації описано в спеціальній літературі.
3.6. Спеціальні види перегонки
Розділення компонентів з близькими температурами кипіння, відносні леткості a яких наближені до одиниці, пов’язане із значними труднощами. Під час роботи з такими сумішами лінія рівноваги настільки зближується з діагоналлю діаграми у – х, що для їх розділення є необхідною дуже велика кількість одиниць перенесення. Величина a може бути збільшеною і розділення полегшеним за зміни тиску перегонки (ректифікація під вакуумом).
Граничним випадком сумішей, що мають близькі температури кипіння, є нероздільно киплячі, або азеотропні, суміші, для яких a = 1. Щоб розділити ці суміші, необхідно значно змінювати тиск, що пов’язано з ускладненням і здорожчанням установки. Дорогим способом розділення азеотропних сумішей також є молекулярна дистиляція (див. нижче).
У багатьох випадках ефективнішими є методи розділення азеотропних сумішей, засновані на введенні в суміш додаткового компонента, так званого розділювального агента, що володіє виборчою дією. При його додаванні леткість і коефіцієнт активності для низькокиплячого компонента зростають значно більше, ніж для висококиплячого, що і полегшує розділення суміші. Застосовуючи різні розділювальні агенти і підбираючи їх концентрацію, можна змінювати в широких межах відносну леткість компонентів початкової суміші і відповідно розподіл її компонентів між рідиною і парою. Здійснення процесів ректифікації у присутності розділювальних агентів є загальною ознакою методів екстрактивної і азеотропної ректифікації. Разом з тим, ці методи істотно відрізняються один від одного. Під час азеотропної ректифікації розділювальний агент утворює азеотропну суміш з одним або декількома компонентами початкової суміші, у вигляді якої він відганяється з ректифікаційної колони як дистилят. Під час екстрактивної ректифікації розділювальний агент повинен мати значно меншу леткість, ніж компоненти початкової суміші і не утворювати з ними азеотропних сумішей. Він відводиться з колони з кубовим залишком.
3.6.1. Екстрактивна ректифікація
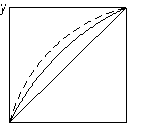
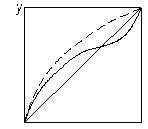
На рис. 3.25 показано вплив додавання розділювального компонента на зміну відносної леткості компонентів бінарної суміші. Пунктиром зображено криві рівноваги, одержані під час розділення суміші близькокиплячих компонентів (рис. 3.25 а) і азеотропної суміші (рис. 3.25 б) у присутності третього компонента. З діаграми у – х видно, що внаслідок різкого підвищення відносної леткості процес розділення значно полегшується, його можна здійснити за меншої кількості ступенів розділення.
а |
б |
Рис. 3.25. Вплив добавки розділювального компонента під час екстрактивної ректифікації: а – суміш близькокиплячих компонентів; б – азеотропна суміш
У схемі установки для екстрактивної ректифікації (рис. 3.26) початкову суміш, що складається з компонентів А+В, подають на живлячу тарілку екстрактивно-ректифікаційної колони 1, яка зрошується згори розділювальним агентом С, більш висококиплячим, ніж компоненти А і В. Компонент В добре розчинний в С, тоді як компоненти А і С взаємно нерозчинні або погано розчинні один в одному. У результаті компонент С екстрагує компонент В (більш висококиплячій у цій суміші) з рідкої та парової фази. Суміш В+С видаляється у вигляді залишку, а дистилят є чистим компонентом А.
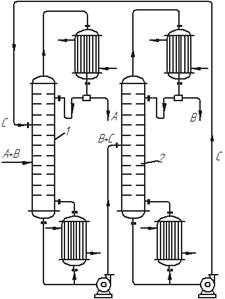
Рис. 3.26. Схема установки для екстрактивної ректифікації:
1 – колона екстрактивно-ректифікаційна; 2 – ректифікаційна колона
для регенерації розділювального агента
За процесом екстрактивної ректифікації йде процес розділення суміші компонентів В+С в ректифікаційні колоні 2. У цій колоні дистилятом є компонент В, більш леткий, ніж С. Регенерований розділювальний компонент С видаляється знизу колони 2 та надходить у колону 1 для повторного використання. Отже, додавання екстрагуючого компонента та розчинення в ньому компонента вихідної суміші ускладнює установку.
Типовим прикладом застосування екстрактивної ректифікації є розділення суміші близькокиплячих бензолу (компонент А) іциклогексану (компонент В) із застосуванням фенолу (компонент С) як розділювального агента. Екстрактивна ректифікація здійснюється тільки безперервним способом.
3.6.2. Азеотропна ректифікація
Під час азеотропної ректифікації зазвичай використовують розділяючий компонент С, який утворює з одним з компонентів початкової суміші (А або В) азеотропну суміш, що має мінімальну температуру кипіння. Утворена більш летка, ніж початкова, азеотропна суміш відганяється як дистилят, а інший, практично чистий, компонент видаляється у вигляді залишку.
Іноді можна підібрати розділювальний агент, що створює з одним з компонентів початкової суміші нову азеотропну суміш з максимальною температурою кипіння. У цьому разі нова азеотропна суміш видаляється у вигляді залишку, а згори колони відбирають дистилят, чистий інший компонент початкової суміші. Можна також здійснити азеотропну ректифікацію за допомогою розділювального компонента, що створює азеотропні суміші з обома компонентами. При цьому співвідношення компонентів А і В у потрійній азеотропній суміші повинно бути іншим, ніж в початковій суміші, що надходить на розділення. За цим варіантом процесу дистилят, що видаляється з колони, є леткою азеотропною сумішшю (з трьох компонентів), а залишок – один з компонентів початкової суміші практично у чистому вигляді.
Підбір розділювального агента здійснюють на основі другого закону Вревського, що вказує напрям зміни складу азеотропної суміші з температурою.
В установці для азеотропної ректифікації (рис. 3.27), що здійснюється з утворенням азеотропної суміші, яка має мінімальну температуру кипіння, вихідна азеотропна суміш (А + В) надходить на живлячу тарілку колони 1, яка зрошується зверху розділювальним агентом С. Згори колони видаляється азеотропна суміш компонентів А + С з мінімальною температурою кипіння (дистилят), знизу колони виходить компонент В (залишок).
На рис. 3.27 показано варіант процесу азеотропної ректифікації, коли азеотропна суміш, що утворюється, складається з компонентів із різною взаємною розчинністю за різних температур.
У цьому разі компоненти А і С, що знаходяться у рідкому стані, є практично взаємно нерозчинні. Тому дистилят після охолоджування розділяється на компоненти А і С у відстійнику 2. Компонент А є кінцевим продуктом, а регенерований компонент С після нагрівання в підігрівачі 3 повертається на зрошування колони 1. У схемі, показаній на рис. 3.27, в дефлегматорі колони 1 конденсується тільки частина пари (А + С), що є необхідною для отримання флегми, а решта зріджується і охолоджується в холодильнику-конденсаторі перед надходженням у відстійник 2.
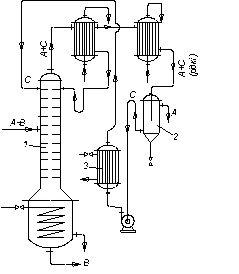
Рис. 3.27. Схема установки для азеотропної ректифікації:
1 – ректифікаційна колона; 2 – відстійник (сепаратор); 3 – підігрівач
Як приклад застосування азеотропної ректифікації можна вказати на процес розділення азеотропної суміші “етиловий спирт – вода” (температура кипіння
~ 78°С), де як розділювальний компонент використовують бензол, що створює з водою і спиртом потрійну азеотропну суміш з мінімальною температурою кипіння (~ 64,8°С). Залишок, що видаляється з колони, є безводним етиловим спиртом.
Процеси азеотропної ректифікації здійснюють безперервним і періодичним способами, причому в останньому випадку компонент, який розділяють, повністю завантажується в куб колони разом з початковою сумішшю, що спрощує схему установки.
Під час азеотропної ректифікації зазвичай є необхідною більша витрата тепла, ніж під час екстрактивної ректифікації. Крім цього, під час азеотропної ректифікації складніше підібрати розділювальний агент і обмеженою є можливість зміни співвідношення його кількості і кількості вихідної суміші порівняно з екстрактивною ректифікацією.
Як розділювальний агент під час екстрактивної і азеотропної ректифікації більшого поширення набувають розчинні тверді речовини, зокрема солі, у присутності яких в необхідний бік змінюється співвідношення компонентів, що розділяються, за фазової рівноваги.
Рідини, що практично не змішуються і частково змішуються, в хімічній технології розділяють гетерогенною азеотропною ректифікацією (рис. 3.28). Наприклад, так вилучають органічні речовини від невеликих домішок розчиненої в них вологи. Процес здійснюють у вичерпних колонах 1 і 2. Початкова суміш, що складається з компонентів А і В, надходить у відстійник 3, де змішується з конденсатом з дефлегматора 4, який є загальним для обох колон. У відстійнику цей конденсат розшаровується на два шари, склади яких відповідають взаємній розчинності компонентів.
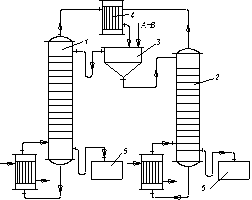
Рис. 3.28. Схема двоколонної установки для гетероазеотропної ректифікації
бінарних рідин, що розшаровуються: 1,2 – ректифікаційні колони;
3 – відстійник; 4 – дефлегматор; 5,6 – збірники
Розчин А і В (верхній шар) зливається в колону 1; тут в результаті перегонки утворюється пара, що є гетероазеотропом, який надходить з дефлегматора 4 у відстійник 3. Залишок з колони 1 є практично чистим компонентом В, який зливається в збірку 5.
Розчин В і А (нижній шар) розділяється в колоні 2 на гетероазеотроп і компонент А. Гетероазеотропблизький за складом до гетероазеотропу, що відгониться з колони 1, тому він прямує через загальний дефлегматор 4 у відстійник 3. Знизу колони 2 відводиться в збірник 6 практично вільний від домішок компонент А.
3.6.3. Молекулярна дистиляція
Вище було розглянуто плівкову ректифікацію під вакуумом, за допомогою якої розділяють нестійкі органічні сполуки. Проте температура кипіння багатьох високомолекулярних речовин (з молекулярною вагою ³300) навіть за значного вакууму залишається дуже високою для того, щоб їх можна було розділити, не побоюючись розкладання. Крім того, для багатьох сумішей необхідно звести до мінімуму тривалість розділення.
Такі суміші розділяють, створюючи дуже глибокий вакуум над поверхнею рідини, що відповідає залишковому тиску 10-3–10-4 мм рт. ст. В умовах глибокого вакууму із зменшенням густини газу зростає довжина вільного пробігу молекул і за достатньо низького залишкового тиску вона може стати більшою від відстані між поверхнями випаровування і конденсації. При цьому велика частина молекул, що відриваються з поверхні випаровування, потрапляє на поверхню конденсації і не повертається з цієї поверхні. Процес здійснюється за наявності близько розміщених поверхонь випаровування і конденсації.
Процес молекулярної дистиляції відбувається випаровуванням рідини з її поверхні за відсутності кипіння. Тому, на відміну від ректифікації, молекулярна дистиляція не характеризується деякими сталими температурою і тиском.
Під час молекулярної дистиляції молекули пари видаляються з поверхні випаровування зразу ж після їх утворення, і рівновага між парою і рідиною не встигає встановитися. Тому розділювальний ефект молекулярної дистиляції визначається не відношенням тиску насиченої пари компонентів суміші або відносною леткістю a, а відношенням швидкостей випаровування компонентів суміші, або коефіцієнтом розділення aм.
Швидкість випаровування 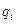 будь-якого компонента ідеального розчину є пропорційною до його мольної частки
будь-якого компонента ідеального розчину є пропорційною до його мольної частки 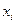 в рідині. Згідно з молекулярно-кінетичною теорією газів
в рідині. Згідно з молекулярно-кінетичною теорією газів
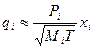 , (3.27)
, (3.27)
де 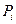 – тиск насиченої пари чистого компонента за температури кипіння суміші; Mi – молекулярна вага компонента; Т – абсолютна температура.
– тиск насиченої пари чистого компонента за температури кипіння суміші; Mi – молекулярна вага компонента; Т – абсолютна температура.
Отже, для бінарної суміші компонентів 1 і 2 можна записати:
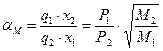 . (3.28)
. (3.28)
Згідно з цим виразом, ступінь розділення під час молекулярної дистиляції є більшою, ніж за рівноважною в 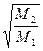 разів.
разів.
Процес молекулярної дистиляції складається з дифузії молекул переважно НК з глибини шару (плівки) рідини до поверхні випаровування, переміщення молекул пари на поверхню конденсації і їх конденсації на цій поверхні. В умовах звичайної дистиляції рідина інтенсивно перемішується під час кипіння з утворенням бульбашок, що піднімаються, і концентрації компонентів вирівнюються в об’ємі рідини. За молекулярної дистиляції швидкість випаровування компонента є пропорційною до його концентрації в рідині (за інших однакових умов). Тому зменшення концентрації компонента в рідині спричиняє зменшення швидкості його випаровування і погіршення розділення. Отже, ефективність процесу залежить від співвідношення швидкостей дифузії (в рідкій фазі) і випаровування компонента. Зазвичай дифузія компонента в рідині є повільнішим процесом, і молекулярну дистиляцію треба здійснювати в умовах, що сприяють прискоренню цієї лімітуючої стадії.
Як відомо з розділу I, швидкість дифузії в рідині можна збільшити за зростання швидкості руху і турбулізації шару рідини, а також за зменшення його товщини. Ці умови створюють в апаратах для молекулярної дистиляції (див. нижче).
Для оцінки ступеня розділення за молекулярної дистиляції використовують поняття про теоретичну молекулярну тарілку (ТМТ), що відповідає ступеню розділення, за якого співвідношення мольних концентрацій компонентів в дистиляті дорівнює відношенню швидкостей випаровування компонентів за повного перемішування шару дистильованої рідини. У виробничих умовах за разового випаровування ступінь розділення коливається від 0,3 до 0,95 ТМТ.
В установці для молекулярної дистиляції (рис. 3.29) на початку процесу необхідно видалити з вихідної суміші розчинені в ній гази і швидко відкачати їх з апарата. Початкова суміш зі сховища 1 надходить в багатоступінчатий дегазатор 2, де у разі нагрівання до 70–100 °С відбувається виділення з неї газів. Гази відкачуються форвакуум-насосами 3 (з перших ступенів) і спареними з ними дифузійно-конденсаційними насосами 4 (з останніх ступенів). Після цього суміш надходить або безпосередньо, або через підігрівач в дистиляційний апарат 5, з якого гази і пару відкачують спареними насосами 3 і 4. Дистилят і залишок прямуютьв окремі збірники 6. Між апаратом і насосами іноді встановлюють пастки 7, охолоджувані холодоагентом, щоб запобігти потраплянню в насоси пари води і органічних рідин.
Для молекулярної дистиляції застосовують плівкові апарати різних конструкцій, описані в спеціальній літературі.
На рис. 3.30 показано промисловий одноступінчастий відцентровий апарат (з плівкою, що піднімається) для молекулярної дистиляції. В корпусі 1 обертається алюмінієвий ротор-випарник 2 конічної форми, що обігрівається зовні електричним нагрівачем 3. Швидкість обертання ротора становить приблизно
400 хв-1. Усередині ротора знаходиться охолоджуваний зсередини гарячої водою конденсатор 5, виготовлений у вигляді розміщених віялоподібних плоских порожнистих елементів. Відстань між внутрішньою поверхнею ротора 2 і поверхнею конденсатора 5 становить 20–30 мм.
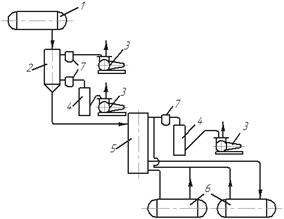
Рис. 3.29. Схема установки для молекулярної дистиляції: 1 – сховище початкової суміші; 2 – дегазатор; 3 – форвакуум-насоси; 4 – дифузійно-конденсаційні насоси;
5 – дистиляційний апарат; 6 – збірники; 7 – пастки
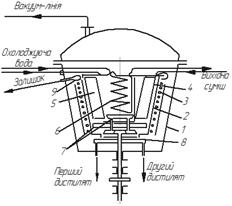
Рис. 3.30. Одноступінчастий відцентровий апарат (з плівкою, що піднімається)
для молекулярної дистиляції: 1 – корпус; 2 – ротор-випарник; 3 – електричний нагрівач; 4 – труба для подавання вихідної суміші; 5 – конденсатор;
6 – охолоджуваний змійовик; 7 – піддон для збору дистиляту; 8 – кільцевий жолоб
для відведення дистиляту; 9 – жолоб для відведення залишку
Рідина, що зазвичай є багатокомпонентною сумішшю, трубою 4 надходить на дно ротора. Під дією відцентрової сили ця рідина у вигляді тонкої плівки турбулентно піднімається вгору нагрітою поверхнею ротора і випаровується у міру піднімання. Пара менш летких компонентів конденсується на поверхні конденсатора 5, а пара більш летких – на поверхні конденсатора-змійовика 6, що охолоджується холодною водою. Рідина стікає в піддони 7, встановлені під конденсаторами, а з них – в кільцеві жолоби 8, звідки трубками видаляються роздільно дві фракції дистиляту. Залишок переливається через верхній край ротора в жолоб 9 і відводиться з апарата.
Молекулярна дистиляція є дорогим способом розділення. Її застосовують у виробництвах деяких пластмас, вітамінів, масел і мастил, жирних кислот, ефірів і ін.
3.6.4. Низькотемпературна ректифікація
Розділення зріджених газових сумішей ректифікацією здійснюють за дуже низьких температур під надлишковим тиском в апаратах, дещо відмінних від звичайних. При цьому продукти розділення одержують повністю або частково у вигляді пари. Проте основні закономірності процесу розділення і методика розрахунку ректифікаційних апаратів є подібними до розглянутих вище.
Відзначимо специфічні особливості пристрою розділювальних апаратів для газових сумішей на прикладі ректифікації рідкого повітря, одержуваного методами глибокого охолоджування. Розділення повітря здійснюють в одноколонних розділювальних апаратах, або в апаратах одинарної ректифікації, двоколонних апаратах, або в апаратах подвійної ректифікації.
Установки одинарної ректифікації. Стиснене в компресорі повітря після очищення від пилу, двоокису вуглецю і водяної пари надходить в теплообмінник 1 (рис. 3.31), де охолоджується продуктами ректифікації (киснем і азотом). Потім повітря надходить в змійовик кип’ятильника 2 колони, девін частково конденсується, віддаючи тепло рідкому кисню, що кипить ззовні змійовика. Пара практично чистого кисню відводиться з кип’ятильника в теплообмінник 1.
Частково сконденсоване повітря, що пройшло через дросельний вентиль 3, ще більше охолоджується. Суміш рідкого і пароподібного повітря надходить на верхню тарілку ректифікаційної колони 4. На тарілках колони відбувається звичайний процес ректифікації: під час багатократної взаємодії стікаючої рідини з парою, що піднімаються знизу, і з якої конденсується кисень (висококиплячий компонент), а з рідини випаровується азот (низькокиплячий компонент). У результаті з верхньої частини колони видаляється пара азоту, що приблизно є рівноважною з повітрям, яке подають в колону і яке містить домішки кисню (не більше 7 – 10 %). До кип’ятильника колони надходить чистий кисень. Як вказувалося, кисень і технічний азот прямують в теплообмінник 1 для охолоджування стисненого в компресорі повітря.
Особливістю пристрою ректифікаційної колони 4 є те, що вона не має дефлегматора і працює як вичерпна колона. Це пояснюється тим, що практично неможливо підібрати охолоджувальний агент для конденсації пари дистиляту (азоту), оскільки для цієї мети знадобилася б рідина, що має температуру, нижчу від температури рідкого азоту. Крім того, як початкова суміш і флегма в колону надходить повітря з дуже низькою температурою, за якої точка перетину робочих ліній може практично відповідати складу дистиляту, що взагалі усуває потребу в дефлегматорі.
Істотним недоліком одинарної ректифікації є втрати кисню з азотом. Приблизно третина кисню видаляється з азотом, забруднюючи його, і тільки дві третини кисню, який знаходиться в повітрі і стискається в компресорі, корисно використовується.
Принципово можливим способом підвищення ступеня чистоти азоту і збільшення виходу кисню під час розділення повітря є живлення ректифікаційної колони початковою сумішшю, багатшою на азот, ніж звичайне повітря. Цей принцип використовують в установках подвійної ректифікації для розділення повітря.

Рис. 3.31. Установка одинарної ректифікації для розділення рідкого повітря:
1 – теплообмінник; 2 – змійовик-кип’ятильник; 3 – дросельный вентиль;
4 – ректифікаційна колона
Установки подвійної ректифікації. У такій установці (рис. 3.32) для попереднього збагачення повітря використовують додаткову нижню колону 1, що працює під високим тиском, більшим, ніж тиск в основній верхній колоні 2, яку встановлюють безпосередньо на колоні 1. Завдяки вищому тиску в нижній колоні вона має дефлегматор (охолоджуваний рідким киснем, що стікає з колони 2), який одночасно є кип’ятильником дляколони 2. Вихідне очищене і охолоджене повітря, стиснене до ~7 aт, вводять в змійовик 3 кип’ятильника колони 1. Віддаючитепло, необхідне для кипіння рідини в кип’ятильнику, повітря конденсується. Зріджене повітря проходить через дросельний вентиль 5, охолоджується ще більше і надходить на живлячу тарілку колони 1, в якій підтримується тиск, що дорівнює 6 aт. У результаті в колоні 1 збирається рідина, збагачена ВК (киснем), і в кип’ятильник 4 стікає рідина, що містить приблизно 40–60 % О2. Тут вона частково випаровується унаслідок теплообміну з повітрям, що проходить змійовиком 3. Пара, що утворилася, піднімається вгору і, взаємодіючи із стікаючою рідиною, збагачується азотом. Пара азоту, що містить 94–96 % N2, надходить в трубки дефлегматора, де вона повністю конденсується і віддає тепло рідкому кисню, що стікає колоною 2, і кисню, що кипить в міжтрубному просторі дефлегматора.
Для здійснення процесу теплообміну в дефлегматорі температура кипіння НК (азоту) в трубках дефлегматора повинна бути вищою від температури кипіння кисню в кип’ятильнику колони 2. Це досягається у разі вказаного вище підвищення тиску до ~ 6 am в колоні 1 порівняно з тиском в колоні 2, що дорівнює ~ 1,5 am.
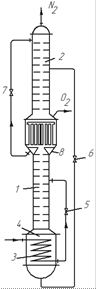
|
| Рис. 3.32.Установка подвійної ректифікації для розділення повітря: 1 – нижня ректифікаційна колона; 2 – верхня ректифікаційна колона; 3 – змійовик; 4 – кип’ятильник нижньої колони; 5–7 – дросельні вентилі; 8 – кишеня |
Збагачена киснем рідина з кип’ятильника нижньої колони надходить через дросельний вентиль 6 (знижуючий її тиск до ~1,5 am) на тарілку живлення верхньої колони 2.
Рідкий азот (з концентрацією 98 % N2), сконденсований в дефлегматорі, ділять на дві частини. Майже половину його кількості подають на зрошування колони 1 для повнішого очищення кисню від азоту, а решта його частини, що збирається в кишені 8, через дросельний вентиль надходить як флегма на зрошування колони 2.
Одержувані азот і кисень містять деяку кількість аргону та інших рідкісних газів, які знаходяться у вихідному повітрі. Для підвищення ступеня чистоти кінцевих продуктів розділення доводиться видаляти частину пари з тієї тарілки колони 1, на якій в найбільшій кількості нагро-
маджується аргон. Подальше розділення рідкісних газів здійснюється низькотемпературною ректифікацією в окремих колонних апаратах.
З верхньої частини колони 2 виводять пару азоту, що містить 99,8–99,9 % N2, знизу колони 1 – технічний рідкий кисень (99,3 % О2).
Відомо також інший спосіб підвищення ступеня чистоти азоту у разі використання апарату, в якому замість додаткової колони задіюють дефлегматор з довгими трубками. Охолоджене повітря з компресора частково конденсується і збагачується азотом. Таке попереднє розділення повітря в дефлегматорі, що працює за вищого тиску, ніж колона 2, дає змогу замінити ним колону 1.
Продуктивність існуючих установок для розділення повітря становить до 7500 м3/год повітря і більше.
Питання до розділу ІІІ
1. Загальні відомості про процес перегонки і ректифікації.
2. Характеристика двофазних систем “рідина – рідина”.
3. Ідеальні суміші. Закон Рауля.
4. Проста перегонка. Види перегонки.
5. Ректифікація. Принципи ректифікації. Схема і принцип роботи безперервно діючої ректифікаційної установки.
6. Класифікація бінарних систем ідеальної і реальної суміші. Рівноважний стан.
7. Схема і принцип роботи періодично діючої ректифікаційної колони.
8. Періодична ректифікація бінарних систем, тарілчасті колони.
9. Рівноважна і робочі лінії процесу ректифікації. Побудова робочих ліній.
10. Матеріальний баланс періодично діючої ректифікаційної колони.
11. Тепловий баланс процесу ректифікації.
12. Залежність між флегмовим числом, витратою теплової енергії на процес і розмірами колони.
13. Конструкція ректифікаційних колон. Загальні відомості.
14. Ректифікація багатокомпонентних сумішей.
15. Спеціальні методи перегонки. Азеотропна і екстракційна ректифікації.
16. Молекулярна дистиляція.
17. Порядок розрахунку тарілчастих і насадкових ректифікаційних колон.
18. Фазова рівновага. Правило фаз.
19. Ідеальні суміші.
20. Р – х діаграма ідеальних сумішей.
21. Діаграма t – х – у (залежність температур кипіння і конденсації від складу фаз).
22. Діаграми рівноваги “пара – рідина” (у – х діаграма).
23. Р – х діаграма для сумішей з позитивним відхиленням від закону Рауля.
24. Фазові діаграми азеотропних сумішей.
25. Суміші взаємно нерозчинних рідин.
26. Фракційна перегонка.
27. Проста перегонка з дефлегмацією.
28. Перегонка з водяною парою.
29. Безперервно діюча ректифікаційна установка.
30. Правило Трутона.
31. Розрахунок мінімального флегмового числа.
32. Розрахунок реального флегмового числа.
33. Періодична ректифікація бінарних сумішей.
34. Складові частини ректифікаційної установки.
35. Барботажні колони.
36. Насадкові колони.
37. Плівкові колони.
38. Розрахунок насадкових колон.
39. Розрахунок тарілчастих колон.
40. Розрахунок ректифікації бінарних систем за допомогою ентальпійної діаграми.
41. Низькотемпературна ректифікація.
42. Установки подвійної ректифікації.
Дата добавления: 2015-07-24; просмотров: 4341;