Общие признаки
Определение. Лизосомы представляют собой цитоплазматические органеллы, в кислой среде которых содержатся многочисленные ферменты, гидролизующие большинство биологических макромолекул (рис. 316-1). Первичные лизосомы представляют собой особые тельца, образующиеся из пластичного комплекса (аппарат Гольджи). Они могут сливаться с другими окруженными мембраной пузырьками, формируя вторичные лизосомы. Последние содержат материал, попавший в клетку извне в результате эндоцитоза, или внутриклеточный материал, поглощаемый в процессе аутофагии. Основная функция лизосом заключается в разрушении использованных макромолекул по ходу их нормального кругооборота и тканевой перестройки. Исследования метаболизма витамина В 12, липопротеинов, пептидных гормонов и факторов роста свидетельствуют о роли лизосом в поглощении этих молекул путем адсорбтивного эндоцитоза. Начальная клеточная вакуоль, образующаяся при адсорбтивном эндоцитозе (рецептосома, или эндосома) сливается с лизосомами. Лизосомные ферменты представляют собой гликопротеины, синтезирующиеся в эндоплазматической сети. Исходные продукты белкового синтеза подвергаются существенным изменениям, в том числе протеолитическому отщеплению, присоединению комплекса олигосахаридов, синтезу маркеров распознавания (в некоторых случаях маннозо-6-фосфата) и компартментализации в первичные лизосомы. Эти процессы протекают в эндоплазматической сети, пластинчатом комплексе и, вероятно, в первичных, если не вторичных, лизосомах.
Концепция лизосомных болезней накопления сложилась в результате изучения гликогеноза II типа (Помпе). Факт накопления гликогена в лизосомах вследствие недостаточности -глюкозидазы, а также данные, полученные при исследовании других аномалий, позволили Эру определить врожденную лизосомную болезнь как такое состояние, при котором: 1) определяется недостаточность какого-либо одного лизосомного фермента и 2) внутри связанных с лизосомами вакуолей появляются необычные отложения (субстраст). Это определение можно видоизменить, включив в него дефекты одиночных генов, влияющие на один лизосомный фермент или более, и тем самым распространить на такие болезни, как муколипидозы и множественная сульфатазная недостаточность. Определение можно расширить и далее с тем, чтобы оно распространялось на недостаточность и других белков, необходимых для функционирования лизосом (активирующие ферменты разрушения сфинголипидов). Данные биохимических и генетических исследований свидетельствуют о том, что эти активирующие белки принимают участие в гидролизе некоторых субстратов.
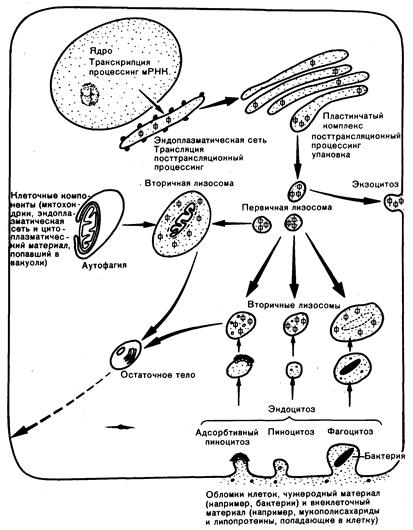
Рис.316-1. Биология лизосом;
Лизосомные ферменты, в том числе их предшественники (Ф), синтезируются в эндоплазматической сети и затем подвергаются посттрансляционному процессингу, который заключается в их упаковке в первичные лизосомы. Первичные лизосомы могут затем вступать в любой из указанных путей.
Лизосомные болезни накопления объединяют большинство болезней накопления липидов, мукополисахаридозы, муколипидозы, болезни накопления гликопротеинов и другие, перечисленные в табл. 316-1. Недостаточность ферментов имеет аутосомно-рецессивную основу, за исключением мукополисахаридоза II (МПС II) Хантера, который наследуется как сцепленный с Х-хромосомой рецессивный признак, и болезни Фабри, которая сцеплена с Х-хромосомой и часто проявляется у женщин. Органами-мишенями оказываются обычные места разрушения той или иной макромолекулы. Например, у лиц с нарушением процесса разрушения миелина в процесс вовлекается белое вещество головного мозга, при нарушении процесса разрушения гликолипидов стромы эритроцитов развивается гепатоспленомегалия, а при нарушении процесса разрушения вездесущих мукополисахаридов — генерализованное повреждение тканей. Накапливающийся материал часто вызывает висцеромегалию или макроцефалию, но может развиться и вторичная атрофия, особенно мозга и мышц. Вообще симптоматика соответствующих болезней обусловливается повреждающим действием накапливающихся веществ, но часто неясно, каким именно образом они вызывают гибель или дисфункцию клеток. Все эти болезни прогрессируют, и многие из них заканчиваются смертью в детском или юношеском возрасте. Для окончательного диагноза наиболее важны результаты определения конкретных ферментов в сыворотке, лейкоцитах или культивируемых фибробластах кожи; соответствующие тесты выбирают, исходя из клиники заболевания. Эти болезни имеют широкие фенотипические колебания, причем многие из них связаны с возрастом, т. е. различают инфантильные, ювенильные и взрослые их формы. Кроме того, при болезнях, обусловленных дефектом одиночного гена, возможны различные сочетания висцеральных, костных и неврологических аномалий.
Таблица 316-1. Лизосомные болезни накопления
| Болезнь | Гетерогенность (возраст начала) | Дефицитный фермент | Накапливающиеся вещества | Неврологические аномалии | Увеличение печени и/или селезенки | Дисплазия костей | Офтальмологические нарушения | Гематологические нарушения | Наследование | Особенности проявления | Ссылки | |||||||||||||||||||||||||||||||||||||||||||||||||
| Ганглиозидоз gmi | Инфантильная (у новорожденного), ювенильная (возраст 6— 20 мес) и взрослая формы | -Галактозидаза | Ганглиозид gmi, гликопротеины, кератансульфат | Отсталость психического развития, судорожные припадки, слепота; при ювенильной форме проявляются позже, при взрослой форме варьируют | Резко выражено В меньшей степени при ювенильной форме, при взрослой форме варьирует | Резко выражена Варьирует при ювенильной и взрослой формах | Вишнево-красные пятна у 50% больных с ювенильной формой, помутнение роговицы варьирует, но более выражено при взрослой форме | Пенистые клетки Вакуолизированные лимфоциты | Аутосомно-рецессивное (АР) | Грубые черты лица, отеки, макэ роглоссия, мукополиса-харидурия, ранняя слепота при инфантильной форме; менее выраженные проявления при ювенильной форме; при взрослой форме часто спондило- | Hers, Van Hoof, гл. 12, Stanbury et al., гл. 46, Но et al. | |||||||||||||||||||||||||||||||||||||||||||||||||
| Синдром Тея—Сакса с вариантами, ганглио- ЗИДОЗ См2 | Инфантильная (возраст 3— 6 мес), ювенильная и взрослая формы | Гексоза-минидаза А | Ганглио- ЗИД См2 | Отсталость психического развития, судорожные приступы, слепота; при ювенильной форме проявляются позднее | Не определяется | Отсутствует | Вишнево-красные пятна при инфантильной форме, редко при ювенильной | Отсутствуют | АР | эпифизарная дисплазия, мукополиса-харидурия +/-Макроцефалия, резкое обострение слуха при инфантильной форме; особенно распространен среди евреев ашкенази | Hers, Van Hoof гл. 13, Stanbury et al., гл. 46, Но et al. | |||||||||||||||||||||||||||||||||||||||||||||||||
| Синдром Сендхоффа, ганглиози- ДОЗ См2 | Инфантильная форма (возраст 3—б мес) | Гексоза-минидаза А и В | Ганглио- ЗИД См2 Глобозид | Отсталость психического развития, судорожные припадки, слепота | Отсутствуют | Отсутствует | Вишнево-красные пятна | Отсутствуют | АР | Макроцефалия, усиленная реакция на звук, висцеральный гистиоцитоз | Hers, Van Hoof, гл. 14, Stanbury et al., гл. 46, Но et al. | |||||||||||||||||||||||||||||||||||||||||||||||||
| Ганглио- ЗИДОЗ См2, АВ-вариант | Все изменения и признаки те же, что и при синдроме Тея—Сакса, '^Н за исключением того, что первично аномален белковый активатор разрушения ганглиозидов ^^И | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Синдром Краббе, га-лактозилце-рамидный липидоз | Инфантильная форма (возраст 2—б мес), позднее начало | Галакто-зилцер-амид-р-га-лактози-даза | Увеличение отношения галакто-зоцеребро-зид/сульфа-тид | Отсталость психического развития, лей-кодистро-фия; варианты при позднем начале | Отсутствуют | Отсутствует | Атрофия зрительного нерва | Отсутствуют | АР | Резкая возбудимость, повышен уровень белка в спинномозговой жидкости (СМЖ), лихорадочное | Hers, Van Hoof гл. 17, Stanbury et al., гл. 43, Но et al. | |||||||||||||||||||||||||||||||||||||||||||||||||
| состояние, сфероцитар-ная нейропа-тология | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Метахром-ная лейко-дистрофия, сульфатид-ный липидоз | Поздняя инфантильная (возраст 1—4 года), ювенильная (возраст 4 года — 20 лет) и взрослая формы | Арилсульфатаза А (цереб-розидсуль-фатаза) | Галакто-зилсульфа-тиды | Отсталость психического развития, лей-кодистро-фия, при взрослой форме психоз и деменция | То же | То же | Атрофия зрительного нерва, менее выраженная при ювенильной и взрослой формах | То же | АР | Повышение уровня белка в СМЖ, нарушение походки при поздней инфантильной форме, периферическая нейропатия | Hers, Van Hoof, гл. 18, Stanbury et al., гл. 44 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Недостаточность | Все изменения те же, что при метахромной лейкодистрофии, за ]— исключением того, что первичный дефект заключается в недостаточности белкового активатора 'Л | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| белкового активатора распада сфинголипи-да I Синдром Нимана— Пика, сфин-гомиелино-вый липидоз | Инфантильная нейропатическая форма (возраст 1— 4 мес), позднее начало нейропатической формы, висцеральная форма | Сфинго-миелиназа ? У некоторых больных специфические изоферменты | Сфинго-миелин | Отсталость психического развития, атаксия и судорожные припадки при нейропатических формах | Резко выражено | Отсутствует | Макуляр-ная дегенерация и вишнево-красные пятна при нейропатических формах | Характерные пенистые клетки, вакуолизированные лимфоциты | АР | Легочные инфильтраты, буроватый цвет кожи; инфантильная нейропатическая форма распространена среди евреев ашке-нази, гистиоциты цвета морской волны | Hers, Van Hoof, гл. 19, Stanbury et al., гл. 41 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Синдром Гоше, глю-козилцер-амидный липидоз | Инфантильная (возраст 1— 12 мес), ювенильная (возраст 2 года — 6 лет), взрослая формы | р-Глюко-церебрози-даза | Глюкозил-церамид | Отсталость психического развития, спастичность, позднее — вялость, атаксия при ювенильной форме, при взрослой форме — отсутствие неврологической симптоматики | Резко выражено, часто гипер-спления | Средняя степень | Обычно отсутствуют | Характерные пенистые клетки | АР | При взрослой форме повышение уровня кислой фосфатазы, патологические переломы; особенно распространен у евреев ашкинази | Hers, Van Hoof, ГЛ: 16, Stanbury et al., гл. 42, Но et al. | |||||||||||||||||||||||||||||||||||||||||||||||||
| Синдром Фабри, три-гексозилце-радоз | Мужчи-ны-гемизи-готы, женщины-гетерозиготы | а-Галак-тозидаза А | Тригексо-зилцерамид | Болевая нейропатия | Отсутствует | | | Отсутствует | Дистрофия роговицы, сосудистая патология, катаракты | Отсутствуют | Сцепленное с' Х-хромосомой доминантное | Кожная ангиокерато ма, тромбозы сосудов, гипогидроз | Hers, Van — Hoof, гл. 15, Stanbury et al., гл. 45, U/^ /»*- nl но ет ai. | ||||||||||||||||||||||||||||||||||||||||||||||||
| Недостаточность кислой липазы | Инфантильная болезнь Воль-мана (возраст 0—3 мес); при позднем начале — болезнь накопления эфиров холестерина (БНЭХ) | Кислая липаза | Эфиры холестерина, триглицериды | Отсталость психического развития, но выраженная слабее, чем задержка роста при болезни Вольмана; все это не отмечается при БНЭХ | Заметно выражено | | | Отсутствует | Отсутствуют | Пенисты клетки, вакуолизированные лимфоциты | е АР | Кальцино. надпочечников, анемия, рвота и задержка роста при болезни Вольмана; фиброз печени и повышение уровня холестерина в крови при crt^av | з Hers, Van Hoof., гл. 20, Sranbury et al., гл. 39 | ||||||||||||||||||||||||||||||||||||||||||||||||
| Синдром Фарбера, недостаточность цера-мидазы | Инфантильная (возраст 0— 4 мес), редко ювенильная формы | Церами-даза | Церамид | Иногда отсталость психического развития, но она может быть вторичной по отношению к соматическим проявлениям | +/- | | | ? | Легкая степень ма-кулярной дегенерации | Отсутствуют | АР | bUJA Артропатия: подкожные, периартикулярные и висцеральные узелки (липограну-лематоз); повышение уровня белка в СМЖ | Hers, Van Hoof, гл. 24, Stanbury et al., гл. 40, Но et al. | ||||||||||||||||||||||||||||||||||||||||||||||||
| Синдром Помпе, гликогеноз II типа | Инфантильная (возраст 0— б мес), ювенильная и взрослая формы | Кислая мальтаза (а-1,4- и 1.6-глюко-зидаза) | Гликоген | Психическое развитие, вероятно, в пределах нормы | Легкая степень гепатомегалии | Отсутствует | Отсутствуют | То же | АР | При инфантильной форме смертельная, скелетная и сердечная миопатия; при взрослой форме преимущественно скелетная миопатия | См. гл. 313, Hers, Van Hoof, гл. 7, Stanbury et al., ГЛ. 6 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Недостаточность кислой фосфатазы | Инфантильная форма (возраст 0—3 мес) | Кислая фосфатаза | Не выяснены | Отсталость психического развития | Выражено | То же | То же | » » | АР? | Смертельные нарушения (наблюдались члены двух семей) | Hers, Van Hoof, гл. 21, Hirschhorn, Weissmann | |||||||||||||||||||||||||||||||||||||||||||||||||
| Фукозидоз | Инфантильная форма (воз- | а-Фуко-зидаза | Гликопеп-тиды, глико-липиды, | Отсталость психического | Определяется | • | Определяется | Отсутствуют | Вакуолизированные лимфоци- | АР | Грубые черты лица, повышение | Hers, Van Hoof, гл. 11, | ||||||||||||||||||||||||||||||||||||||||||||||||
| раст 3—12 мес), ювенильная форма | Олигоса-хариды | развития | j | ты, пенистые клетки | уровня электролитов в потовой жидкости; ангио-кератома при ювенильной | Но et al., Sranbury et al., гл. 38 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Маннози- ДОЗ | Инфантильная форма (возраст б—18 мес), более легкая форма | а-Ман-нозидаза | Олигоса-хариды | То же | Выражено | То же | Катаракты, помутнение роговицы | Вакуолизированные лимфоциты, гранулированные нейтрофи | АР | форме Грубые черты лица, увеличение размеров языка | Hers, Van Hoof, гл. 11, Stanbury et al., гл. 38 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Аспартил-глюкозамин-урия | Ранняя взрослая форма | Аспар-тилглюко-заминами-даза | Аспартил-глюкозамин, гликопеп-тиды | » » | Отсутствует | » » | Помутнение хрусталика | лы Вакуолизированные лимфоциты | АР | Грубые черты лица; болезнь выявляется при анализе мочи на аминокис | Hers, Van Hoof, гл. 24, Stanbury et al., гл. 38 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Мукопо-лисахаридоз IH и IS | Инфантильный синдром Гурлер (возраст 6—12 мес), промежуточная форма, в зрелом возрасте — синдром Шейе | а-Идуро-нидаза | Дерматан-сульфат, гепарансульфат | Отсталость психического развития, при синдроме Шейе отсутствует | Выражено | | | Резко выражена | Помутнение роговицы | Гранулированные лимфоциты | АР | лоты Грубые черты лица, сердечно-сосудистая патология, анкилоз | Hers, Van Hoof, гл. 8 и 9, Stanbury et al., гл. 31, McKusick | ||||||||||||||||||||||||||||||||||||||||||||||||
| Синдром Хантера, му-кополисаха-ридоз II | Тяжелая инфантильная форма (возраст 6— 12 мес), легкая ювенильная форма | Идуроно-сульфат-сульфатаза | То же | Отсталость психического развития, менее выраженная при легкой форме | » | | | То же | Дегенерация сетчатки; незначительное помутнение роговицы | То же | Сцепленное с Х-хромое омой | То же | |||||||||||||||||||||||||||||||||||||||||||||||||
| Синдром Санфилип-по А, му-кополиса-харидоз IIIA ( | Поздняя инфантиль | Гепаран-N-сульфа-таза (сульфами-даза) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Синдром Санфилип-по В, му-кополиса-харидоз IIIB | ная форма (возраст 1—4 года) | N-Аце-тил-а-глю-козамини-даза | Гепарансульфат | Тяжелая форма отсталости психического развития | Слабо выражено | Слабо выражена | Отсутствует | Гранулированные лимфоциты | АР | Некоторое огрубение черт лица | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Синдром Санфилип-по С, мукополисахаридов те | То же | Ацетил-КоА: а-глюко-заминид-N-ацетил-трансфера-за | | | То же | То же | То же | То же | То же | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Синдром Санфилип-по D, мукополисахаридов HID I | N-Аце-тилглюко-замин-б-сульфат-сульфатаза | | | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Синдром Моркио, мукополисахаридоз IV | Некоторые колебания | N-Аце-тилгалак-тозамин-б-сульфат-сульфатаза | Кератан-сульфат | Отсутствуют | То же | | | Тяжелая степень | Помутнение роговицы | Гранулированные нейтрофилы | АР | Выраженные деформации, гипоплазия зубовидного отростка II шейного позвонка, регургитация аортального клапана | |||||||||||||||||||||||||||||||||||||||||||||||||
| Синдром Марото— Лами, муко-полисахари-доз VI | Колебания тяжести и степени повреждения сердечнососудистой системы | N-Аце-тил-гексо-замин-4-сульфат-сульфатаза (арилсульфатаза В) | Дерматан-сульфат | То же | Определяется | | | Резко выражена | То же | Гранулированные нейтрофилы и лимфоциты | АР | Некоторое огрубение черт лица, анкилоз, поражение клапанов сердца | |||||||||||||||||||||||||||||||||||||||||||||||||
| Недостаточность р-глюкурони-дазы, муко-полисахари-доз VII | Выявлены несколько больных со всеми формами (от инфантильной до взрослой) | р-Глюку-ронидаза | Дерматан-сульфат, гепарансульфат (?) | Отсталость психического развития, отсутствующая (?) у некоторых взрослых больных | Выражено | | | Выражена | Помутнение роговицы | Гранулированные нейтрофилы | АР | Грубые черты лица, патология сосудов | |||||||||||||||||||||||||||||||||||||||||||||||||
| Множест венная суль-фатазная недостаточность | Поздняя инфантильная форма (возраст 1—4 года) | Арилсульфатазы А, В и С, другие сульфатазы | Сульфати-ды, мукополисахариды | Отсталость психического развития | Слабо выражено | j | То же, что при МПС | Дегенерация сетчатки | Вакуолизированные и гранулированные клетки | АР | Ихтиоз, сочетанный фенотип МПС и ме-тахромной лейкодист-рофии | Hers, Van Hoof, гл. 8 и 18, Stanbury et al., гл. 44 | ||||||||||||||||||||||||||||||||||||||||||||||||
| Сиалидоз | Врожденная, инфантильная, | Глико-протеин-нейрамини- | Сиалило-лигосахари-ды | Отсталость психического | Средняя степень, слабее при | • | Выражена, слабее или отсутст- | Вишнево-красное пятно | Вакуолизированные лимфоциты | АР | Фенотип МПС при всех формах, | Hers, Van Hoof, гл. 8, | ||||||||||||||||||||||||||||||||||||||||||||||||
| ювенильная формы, вишнево-красное пятно, мио-клонус | даза (сиа-лидаза) | развития, миоклонус | поздней форме | j | вует при поздней форме | кроме формы с вишнево-красным пятном и миоклонусом | Stanbury et al., гл. 38 | |||||||||||||||||||||||||||||||||||||||||||||||||||||
| Муколи-пидоз II, Y-клеточная болезнь | Инфан- } тильная форма (возраст 0—3 мес) | Гликопротеины, гли-колипиды | Отсталость психического развития | Не всегда определяется | | | Резко выражена | Помутнение роговицы | Вакуолизированные и гранулированные нейтрофи | АР | Грубые черты лица, включения в культивируемых | Hers, Van Hoof, гл. 8, Stanbury et al., | |||||||||||||||||||||||||||||||||||||||||||||||||
| My коли -пидоз III, псевдополи-дистрофия Гурлер | Поздняя инфантильная форма (возраст старше 2 лет) j | УДФ-N- ацетилглю-козамин-(GlcNAc): гликопро-теин-GlcNaCI-фосфо-трансфе-раза | То же | Некоторая отсталость психического развития | Отсутствует | Выражена | То же | лы Вакуолизированные плазматические клетки | АР | фибробластах, мукопо-лисахарид-урия в пределах нормы Грубые черты лица, включения в культивируемых фибробластах, контрактуры суставов, поражение клапанов сердца, му-кополисаха-ридурия в пределах нлпмкт НииДЮ! | гл. 37 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Муколи-пидоз VI | Инфантильная форма | Ганглио-зиднейра-минидаза (?) | Многочисленные | Отсталость психического развития | Отсутствует | | | Отсутствует | Помутнение роговицы, дегенерация сетчатки | Отсутствуют | АР | Диагноз основывается на результатах электронной микроскопии; особенно распространен среди евреев ашкинази (?) | Stanbury et al., гл. 37 | ||||||||||||||||||||||||||||||||||||||||||||||||
| Восковидные липо-фусцинозы нейронов | Поздняя инфантильная, ювенильная и взрослая формы | Неизвестен | «Воск», «липофусцин» | Отсталость психического развития, деменция, варьирующая у взрослых, судорожные припадки | То же | | | То же | Атрофия зрительного нерва, маку-лярная дегенерация, пигментный ретинит | Вакуолизированные лимфоциты Гранулированные нейтрофилы | АР АР | В диагностике помогает электронная микроскопия; степень генетической гетерогенности неизвестна | Hers, Van Hoof, гл. 23 | ||||||||||||||||||||||||||||||||||||||||||||||||
Диагностика. Подозрение на лизосомную болезнь появляется обычно при прогрессирующей дисфункции нервной системы, висцеромегалии, нарушениях скелета или каких-то более специфических аномалиях (см. табл. 316-1). Отличительной особенностью этих болезней служат прогрессирующие или дегенеративные процессы. Дегенеративные процессы у нормально развивающегося ребенка приводят к замедлению развития, а затем уже к утрате им ранее приобретенных навыков. При сборе анамнеза необходимо обращать особое внимание на развитие в детстве, неврологические симптомы, включая судороги, а также нарушения зрения и слуха, физический рост и более специфические показатели, такие как огрубение черт лица, помутнение роговицы, усиление ранних рефлюксов, растяжение живота, боли в суставах, их тугоподвижность, грыжи и рецидивирующие инфекции. При сборе семейного анамнеза в случае аутосомных рецессивных заболеваний можно выявить аналогичные симптомы у сиблингов или других кровных родственников, а в случае Х-сцепленных аномалий обнаружить других больных членов семьи мужского пола. Целесообразно учитывать этническую принадлежность больного, так как ряд болезней накопления липидов чаще регистрируется у евреев ашкенази, а маннозидозы и аспартилглюкозаминурия — у жителей Скандинавии. Ювенильная форма сиалидоза распространена в Японии.
При осмотре можно обнаружить увеличение окружности черепа. В начале развития некоторых мукополисахаридозов и гликогенозов отмечается гигантизм, тогда как более поздним проявлением многих заболеваний служит низкорослость. При офтальмологическом обследовании с помощью щелевой лампы тщательно осматривают глазное дно. Могут увеличиваться размеры языка, черты лица становятся грубыми, присоединяется гепатоспленомегалия. Аномалии скелета могут заключаться в кифозе, ширококостности и тугоподвижности суставов. Изменения кожи встречаются редко, обычно только при фукозидозе, сиалидозе, болезнях Фабри и болезни Гунтера. При тщательном неврологическом обследовании необходимо попытаться определить степень повреждения серого и белого вещества мозга, а также периферических нервов. Предварительные диагностические исследования должны включать изучение мазков периферической крови для выявления вакуолизированных или гранулированных лейкоцитов, пробу на мукополисахариды в пятне мочи и обзорную рентгенографию скелета. Предпочтительный алгоритм диагностики заключается в использовании всех этих методов для выбора фермента, активность которого требует проверки при исследовании сыворотки, лейкоцитов или культуры фибробластов кожи. При положительном скрининг-тесте на мукополисахариды или при убедительных клинических данных можно провести количественный анализ ^на мукополисахариды. При неопределенности результатов обследования целесообразно произвести биопсию кожи, костного мозга, слизистой оболочки прямой кишки, печени, периферического нерва, конъюнктивы или других тканей для проведения световой и электронной микроскопии. Результаты электронно-микроскопического исследования в зависимости от наличия или отсутствия переполненных лизосом могут подтвердить или отвергнуть общее предположение о лизосомной болезни. И в этих случаях определение ферментов — наиболее подходящий способ диагностики. Если получены веские данные в пользу лизосомной болезни накопления, но не выявлена недостаточность какого-либо фермента, целесообразно начать дальнейшие исследования с химического анализа биоптатов печени или мозга.
Гетерогенность. Лизосомные болезни накопления значительно различаются по клиническим и биохимическим признакам. Биохимические и генетические основы этих различий обсуждались в гл. 57 и 305. Коротко говоря, структурный ген лизосомного фермента производит продукты, которые после посттрансляционной модификации превращаются в гликопротеины, но при этом часто образуется несколько вариантов последних с разными электрофоретическими свойствами — изоферменты. Эти изоферменты способны гидролизовать один или несколько субстратов, причем субстратная специфичность разных изоферментов может различаться. Различия в субстратной специфичности обусловлены также присутствием сходных, но генетически разных ферментов, например -галактозидаз. Мутация гена может полностью лишить фермент активности или только снизить ее, изменить способность фермента к посттрансляционной модификации или его активность по отношению к конкретным субстратам.
В большинстве случаев различия в тяжести заболевания у разных больных, равно как и разные сочетания висцеральных, скелетных, неврологических, глазных и других проявлений одной и той же болезни, определяются разными мутациями структурных генов лизосомных ферментов. Гетерогенность еще больше увеличивается из-за рецессивной природы большинства этих болезней, так как у каждого больного должно быть два мутантных гена в одном локусе. Точный характер мутаций в двух копиях одного гена может быть неодинаковым, что делает больного генетически компаунд-гетерозиготой. В этом случае один или оба гена могут кодировать фермент с некоторой остаточной активностью по отношению к одному или нескольким субстратам. Вероятными примерами компаунд-гетерозигот могут служить больные с промежуточными клиническими фенотипами мукополисахаридоза I типа (МПС I). На молекулярном уровне большинство больных с лизосомными болезнями накопления можно, вероятно, отнести к компаунд-гетерозиготам. Несмотря на то что с клинической точки зрения целесообразно различать инфантильный, ювенильный, взрослый, нейропатический или ненейропатический фенотипы болезни, существование разных мутантных аллелей и генетических комбинаций объясняет случаи аберрантности или промежуточности состояний по сравнению с их обычным фенотипом. Другой тип гетерогенности подтверждают МПС III А, В, С и D, представляющие собой весьма сходные болезни, но обусловленные дефектами разных генов. Таким образом, в основе внешней клинической гомогенности может лежать биохимическая гетерогенность.
Еще большую сложность проблеме придает тот факт, что активность некоторых ферментов определяется сочетанием неодинаковых субъединиц молекулы. В результате недостаточность одного и того же фермента может быть обусловлена разными мутациями (например, недостаточность гексаминидазы А при болезнях Тея— Сакса и Сендхоффа), которые объясняют также множественную недостаточность ферментов при дефекте одиночного гена, например при болезни Сендхоффа. Лизосомные болезни накопления могут быть результатом генетических аномалий и на уровне посттрансляционной модификации лизосомных ферментов, равно как и общих дефектов целостности и функции лизосом. Муколипоидозы II и III отражают ситуацию, при которой дефект одиночного гена меняет способность ряда лизосомных ферментов проникать в лизосомы. Таким образом, гетерогенность может обусловливаться и мутациями, не затрагивающими структурные гены самих ферментов. Лучшему пониманию природы фенотипической и генотипической гетерогенности должна способствовать дальнейшая расшифровка биохимической идентичности, структуры субъединиц, посттрансляционного процессинга и субстратной специфичности лизосомных ферментов.
Установлению клинического диагноза способствует, но в то же время в чем-то и осложняет широкое использование синтетических субстратов при определении активности лизосомных ферментов. С помощью таких субстратов зачастую регистрируется активность группы близких, но разных ферментов. Так, при использовании искусственного субстрата активность р-галактозидазы на самом деле может отражать сумму действия разных -галактозидаз, кодируемых разными структурными генами и имеющих разную субстратную специфичность. Для того чтобы придать клиническую значимость результатам этих определений, приходится варьировать условия in vitro для того, чтобы вычленить действие именно того фермента, недостаточность которого характерна для данного заболевания. Однако генетическая гетерогенность проявляется в том, что мутантный фермент либо гидролизует природный, но не искусственный субстрат, либо наоборот. Например, если использовать искусственный субстрат, то у здорового человека можно обнаружить недостаточность гексозаминидазы А, а у больного с синдромом Тея— Сакса — весьма высокую ее активность. Патология коррелирует со способностью фермента гидролизовать только природный субстрат — ганглиозид GM2. Все это очень важно для скрининга гетерозигот и для пренатальной диагностики. Эти феномены требуют не ограничиваться определением активности ферментов в отношении искусственных субстратов, если результаты определений противоречат отчетливым клиническим, электронно-микроскопическим или химическим признакам болезни.
Лечение и профилактика. В настоящее время отсутствуют эффективные специфические средства лечения, поэтому оно остается в основном симптоматическим. Во многих случаях болезнь неумолимо прогрессирует, и облегчить состояние больного невозможно. При болезни Фабри, часто сопровождающейся почечной недостаточностью, помогает пересадка почки, а у взрослых при болезни Гоше — спленэктомия. Большое внимание привлекла возможность заместительной терапии ферментами путем трансплантации органов или фибробластов, инфузии плазмы, лейкоцитов и ферментов в чистом виде или заключенных в эритроциты или липосомы. Несмотря на то что эти мероприятия могли бы смягчить симптоматику, не связанную с повреждением центральной нервной системы, их эффективность остается недоказанной. Наибольшие проблемы при лечении связаны именно с повреждением центральной нервной системы, поскольку гематоэнцефалический барьер служит дополнительным препятствием для проявления эффекта при заместительной ферментной терапии.
Важно при этих болезнях генетическое консультирование. Все лизосомные болезни накопления, при которых известна недостаточность конкретного фермента, могут или могли бы быть диагностированы in utero, поскольку активность лизосомных ферментов экспрессируется в культивируемых клетках амниотической жидкости, равно как и в культуре фибробластов кожи. Для пренатального диагноза можно также прибегать к помощи биопсии ворсинок плаценты. Несмотря на то что при этом несколько повышается частота выкидышей, члены семей с высоким генетическим риском очень заинтересованы в возможности ранней диагностики. Иногда удается выявить гетерозигот среди близких родственников, но обычно трудно получить согласие достаточного для статистического анализа числа лиц. Выявление гетерозигот осложняет также случайная инактивация Х-хромосом у 46 ХХ-носителей болезней, сцепленных с Х-хромосомой, но следует настойчиво проводить генетическое консультирование женщин из группы риска. Более эффективным профилактическим методом служит выявление гетерозигот перед их вступлением в брак и рождением детей. Реальность этого подхода доказана программами выявления гетерозигот по болезни Тея—Сакса. Эти программы способствовали снижению частоты соответствующих болезней, вероятно, в связи с широким тестированием и влиянием на планирование рождения детей супружескими парами из группы риска по рождению больных детей; высокая частота гетерозигот среди евреев ашкенази и доступность биохимических методов выявления носительства гена болезни Тея—Сакса облегчили проведение этой программы. Распространение этого подхода при других болезнях и в популяциях с меньшей частотой гетерозигот требует разработки надежных методов выявления последних. Даже в оптимальных условиях генетические варианты могут обусловливать ложноположительные или ложноотрицательные результаты любой программы скрининга.
Клонирование генов лизосомных ферментов. Сообщается о клонировании ДНК, кодирующих ряд лизосомных ферментов, и в конце концов большинство этих генов будет клонировано. Работа в этом направлении должна обеспечить лучшее понимание биохимии и генетики лизосомных болезней. Основная надежда возлагается на то, что доступность клонированных генов позволит осуществить генную заместительную терапию в той или иной форме.
Дата добавления: 2015-07-22; просмотров: 1210;