КОАГУЛЯЦІЯ КОЛОЇДНИХ ДОМІШОК|нечистот| ВОДИ
Якщо очищення води від важких|тяжких| грубодисперсних домішок|нечистот| може бути здійснене звичайним|звичним| відстоюванням, то виділення колоїдно-дисперсних речовин з|із| води вимагає застосування процесу коагуляції. Під коагуляцією розуміють фізико-хімічний|фізико-хімічний| процес злипання колоїдних часток|частинок| і утворення грубодисперсної макрофази (флокул|) з|із| подальшим|наступним| її виділенням з|із| води.
Колоїдні частки мають вельми малі розміри і тому беруть участь в броунівському русі, в той же час вони володіють помітною швидкістю дифузії (10–1 – 10–3 см2/с), що сприяє вирівнюванню концентрації часток за об'ємом. Колоїдні системи володіють надлишком вільної енергії за рахунок надзвичайно розвиненій питомій поверхні часток. Термодинамічно така система повинна мимоволі прагнути до стану, в якому її вільна енергія була б мінімальна, тобто до мимовільного зменшення поверхні, а отже, і до укрупнення часток. Проте на практиці колоїдні системи володіють вельми високою агрегативною стійкістю. Така стійкість при малих розмірах часток сприяє седиментаційній стійкості (постійності концентрації домішок в об’ємі води), оскільки гравітаційна сила, що викликає седиментацію, нівелюється силами дифузії. Агрегативна стійкість колоїдної системи пояснюється існуванням подвійного електричного шару іонів і стрибка потенціалу на кордоні розділу фаз.
Подвійний електричний шар виникає на поверхні часток|частинок| унаслідок|внаслідок| різних діелектричних властивостей дисперсної фази і середовища|середовища|, дії молекулярних сил, що забезпечують виборчу|вибіркову| сорбцію іонів, або часткової дисоціації поверхневих|зверхніх| молекул речовини часток|частинок|. Іони, що розташовані на поверхні частки|частинки|, називаються потенціалутворюючими|. Унаслідок|внаслідок| появи заряду на частці|частинці| довкола|навколо| неї концентруються іони протилежного знаку заряду – протиіони|. Концентрація протиіонів максимальна| біля|в| поверхні (щільний іонний шар) і убуває із|із| збільшенням відстані від поверхні частки|частинки| (зовнішній дифузний шар). Це пояснюється|тлумачить| одночасною дією як сил тяжіння (електричних і молекулярних), так і дифузійних сил, прагнучих до вирівнювання концентрації| іонів в об'ємі|обсязі| води.
Теоретично дифузний шар іонів поширюється на весь об'єм води, практично ж його товщину визначають дебаєвською довжиною λ.Так, для симетрично-валентного електроліту в припущенні, що один вид іонів сорбується на поверхні частки, а інший складає проти іони
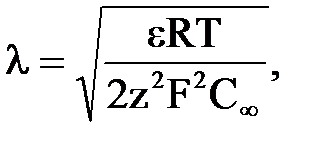
| (2.1) |
де F – постійна Фарадея, рівна 96 487 Кл/моль;
R – універсальна газова постійна, рівна 8,3143 Дж/(моль·К);
z – заряд іонів;
ε – діелектрична проникність середовища|середовища|, Ф/м;
С∞ – концентрація іонів в глибині розчину, моль/л.
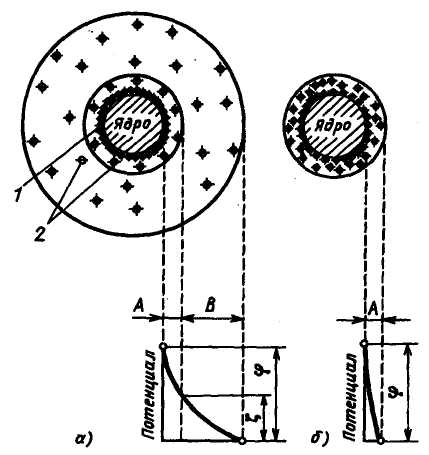
Рис. 2.1. Будова міцели:
а – ξ-потенціал більше нуля; б – ізоелектричний стан: ξ-потенціал дорівнює нулю; 1– потенціалутворюючі іони; 2 – протиіони; А – адсорбційний шар; В – дифузний шар.
Значення величини λ непостійне і, як видно з формули (2.1), залежить від концентрації розчину, заряду іонів, температури. Наприклад, у випадку нейтральної води при 298 К (СН+ = СОН- = 10–7 г-іон/л) дебаєвська довжина λ = 9,6 нм, а у 0,1 молярному розчині одновалентного електроліту λ = 0,96 нм.
Таким чином, навколо частки виникає подвійний електричний шар, що включає потенціалутворюючі іони і протиіони (рис. 2.1). У спокійному стані комплекс частки з подвійним електричним шаром (міцела) електронейтральний.
При русі частка|частинка| захоплює з|із| собою тільки|лише| частину|частину| прилеглого до її поверхні шару води. Зовнішня частина|частина| дифузного шару залишається поза|зовні| сферою дії частки|частинки|. Це призводить|наводить| до появи електрокінетичного потенціалу|, або ζ-потенціалу, між часткою|частинкою| і розчином. Значення ζ-потенціалу| залежить від кількості протиіонів|, що захоплюються з|із| часткою|частинкою|; з|із| їх збільшенням ζ-потенціал зменшується|. Підвищення концентрації протиіонів| у дифузному шарі повинно призводити|наводити| до збільшення їх концентрації в щільному шарі і, отже, до зниження ζ-потенціалу. Більш того|більше того|, підвищення концентрації протиіонів| в дифузному шарі може призвести до перезарядки частки|частинки| (змінюється знак заряду). Природно, що при цьому існує певна концентрація протиіонів|, при якій ζ-потенціал стає| рівним нулю.
Колоїдні частки, що знаходяться в природних водах (пісок, глинисті речовини, гумінові кислоти), в основному набувають заряду за рахунок дисоціації поверхневих молекул. Оскільки ці речовини амфотерні, то вигляд і ступінь їх дисоціації залежать від значення pH розчину. Значення pH, при якому ці речовини не диссоциируют, називається ізоелектричним. При ізоелектричному значенні pH значення ζ-потенціалу дорівнює нулю. Для домішок природних вод значення ізоелектричних pH знаходиться в кислій області. Так, для глини pHи.э = 5, для гумінових речовин pHи.э = 3,5 ÷ 4,5. Оскільки pH природної води зазвичай дорівнює 6,5 ÷ 8,5, то колоїдні домішки дисоціюють як кислоти з придбанням негативного знаку ζ-потенціалу часток щодо розчину. Таким чином, в природній воді основна маса колоїдних часток має однаковий негативний заряд. Крім того, частки глини і гумусу здібні до адсорбції іонів, причому остання знижує їх стійкість до агрегації. Найбільшою мірою знижують стійкість тривалентні іони Fe3+ і Al3+. Іони, що створюють подвійний електричний шар, сприяють утриманню молекул води біля часток і виникненню у зв'язку з цим шару гідрату, що перешкоджає зіткненню часток одна з одною.
Агрегативна стійкість дисперсних систем залежить від характеру сил, що діють між частками|частинками|. На частки|частинки|, що мають однаковий знак заряду, діють одночасно молекулярна сила тяжіння (Ван-дер-Ваальсова сила) і електростатична сила відштовхування.
Молекулярна сила тяжіння двох сферичних часток радіусом r, що знаходяться на відстані R (між їх центрами) одна від одної, описується так:
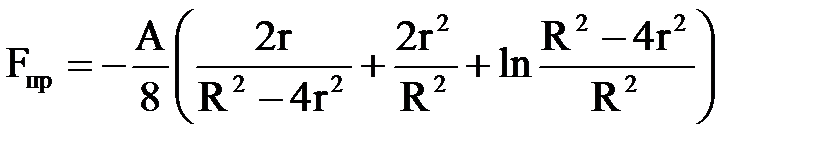
| (2.2) |
де А – константаГамакера, приблизно рівна 10–6 Вт.
При зближенні часток|частинок| до дуже малої відстані ця залежність спрощується:
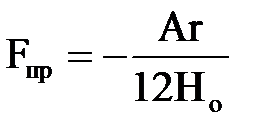
| (2.3) |
де Hо = R – 2r – відстань між крайніми точками часток, що зближуються.
За умови, що Но ≪ r, сила відштовхування таких часток визначається рівнянням Дерягіна–Ландау:
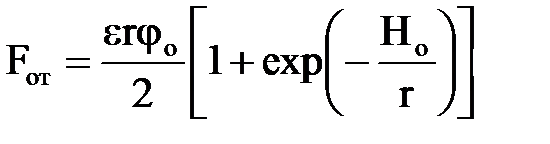
| (2.4) |
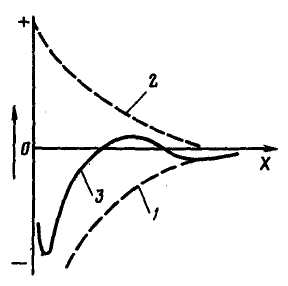
Рис. 2.2. Потенційні криві взаємодії між частками:
1–энергия відштовхування; 2 – енергія тяжіння; 3 – результуюча крива
На рис. 2.2 показаний характер зміни сил тяжіння і відштовхування залежно від відстані між частками. Оскільки закони зміни сил відштовхування і тяжіння різні, результуюча сила має два енергетичні мінімуми (потенційні ями), досягши яких можливе зчеплення часток одна з одною. Правий мінімум забезпечує «далеку» коагуляцію з менш міцним зв'язком між частками. Міцніший зв'язок спостерігається в лівій потенційній ямі («ближня» коагуляція), проте частки, що для цього зближуються, повинні володіти достатньою енергією для подолання енергетичного бар'єру (максимум на результуючій кривої). Подальшому зближенню часток перешкоджають гідратовані шари води на їх поверхні.
У природній воді колоїдні частки|частинки| не володіють достатньою енергією для подолання|здолання| енергетичного бар'єру, а енергія зчеплення в правій потенційній ямі недостатня для їх утримання в комплексі; цим пояснюється|тлумачить| їх підвищена агрегативна| стійкість. Для підвищення можливості|спроможності| зчеплення часток|частинок| необхідно понизити|знизити| силу відштовхування шляхом зменшення значення ζ-потенціалу. Експериментально встановлено|, що при зниженні ζ-потенціалу| до 0,03 В починається|починає| процес зчеплення часток|частинок|, тобто процес коагуляції. Для цього досить ввести|запроваджувати| у воду сильний електроліт, дисоціація якого дозволить збільшити кількість протиіонів| у подвійному електричному шарі з|із| відповідним зниженням значення |ζ -потенціалу|.
Проте|однак| такий процес можливий лише при дуже низьких рН|, що викликає|спричиняє| значні незручності на практиці, які пов'язані із захистом устаткування|обладнання| від корозії, і, крім того, призводить|наводить| до підвищення солевмісту води. Тому при підготовці додаткової води застосовується процес, заснований на взаємній коагуляції колоїдів, для чого у воду вводяться|запроваджують| реагенти, що створюють в ній колоїдний розчин з|із| позитивно зарядженими частками|частинками|. Це порушує стійкість колоїдної| системи і призводить|наводить| до укрупнення часток|частинок|, що створюють її.
В якості реагентів, що називаються коагулянтами, зазвичай застосовують сірчанокислі солі Al2(SO4)3 і FeSO4. Ці солі у воді майже повністю дисоціюють:
| Al2(SO4)3 ⇄ 2Al3+ + 3SO42–; | (2.5) |
| FeSO4 ⇄ Fe2+ + SO42–. | (2.6) |
Катіони слабких основ Al3+ і Fe2+ легко піддаються гідролізу. Так, гідроліз іонів Al3+ протікає таким чином:
| Al3+ + Н2О ⇄Al(ОН)2+ + Н+; | (2.7) |
| Al(ОН)2+ + Н2О ⇄Al(ОН)2+ + Н+; | (2.8) |
| Al(ОН)2+ + Н2О ⇄Al(ОН)3 + Н+, | (2.9) |
а гідроліз іонів Fe2+:
| Fe2+ + H2O ⇄ Fe(ОН)+ + H+; | (2.10) |
| Fe(ОН)+ +H2O ⇄ Fe(ОН)2 + H+. | (2.11) |
У лужному середовищі|середовищі| (рН| > 8) і при достатній кількості кисню гідрат закису заліза окислюється|окисляється| в менш розчинний гідрат його окислу:
| 4Fe(ОН)2 + O2 + 2H2O ⇄4Fe(ОН)3. | (2.12) |
Як видно з наведених реакцій, досить повний гідроліз іонів А13+ і Fe2+ можливий лише за умови відведення іонів Н+. У природній воді зв’язування цих іонів відбувається згідно реакції
| Н+ + НСО3– ⇄Н2СО ⇄СО2 + Н2О, | (2.13) |
тому наявність бікарбонат-іонів у воді є необхідною умовою для забезпечення глибокого протікання процесу гідролізу. При недостатній величині лужності концентрація іонів Н+ може регулюватися введенням NаOH, але це призводить до збільшення солевмісту води. Таким чином, як видно з реакції гідролізу, його глибина істотно залежить від рН води. При рН > 7,5 утворюється тільки Al(ОН)3, при нижчих значеннях рН виходять також Al(ОН)2+ і Al(ОН)2+, які потім, з'єднуючись з сульфат-іонами, утворюють важкорозчинні з'єднання середніх солей алюмінію: Al2(ОН)4SO4 – при рН ближче до 7 і Al(ОН)SO4 – при рН ближче до 5,5. Ці з'єднання в інтервалі значень рН = 5,5 ÷ 7,5 утворюють колоїдний розчин з вельми малим значенням ζ-потенціалу. При рН < 5,5 гідроокис алюмінію розчиняється повністю, а при рН > 8 утворюються іони AlO2–.
Таким чином, при коагуляції води за допомогою Al2(SO4)3 необхідно підтримувати рН в інтервалі 5,5 – 7,5. У воді, що не містить сторонніх іонів, ізоелектричне значення рН для Al(ОН)3 складає 7,6 – 8,2, а при їх наявності зменшується на 1 – 1,5 внаслідок адсорбції іонів на поверхні часток. Утворення Fe(ОН)3 відбувається досить повно і швидко лише при рН > 8, для чого необхідно дозувати у воду разом з коагулянтом луг або поєднувати коагуляцію з процесом вапнування.
Процес коагуляції має дві стадії: приховану і явну. На прихованій стадії відбувається формування колоїдного розчину гідроксидів Al3+ або Fe3+ і утворення мікропластівців. Саме на цій стадії коагуляції вода в основному і очищається від первинних колоїдних домішок. А потім на другій стадії процесу утворюються крупні пластівці (флокули) розміром 1–3 мм, які, володіючи високою сорбційною здатністю, можуть додатково витягувати домішки з води.
При очищенні природних вод в процесі коагуляції одночасно беруть участь домішки|нечистоти| різного ступеня|міри| дисперсності (у тому числі і грубодисперсні) і різної природи (органічні і неорганічні). Такий процес називається гетероадакоагуляцією|. Відмітною його особливістю є|з'являється| переважна залежність процесу від значення заряду часток|частинок|, заряджених слабкіше|слабий|. Такі частки|частинки| можуть злипатися не лише|не тільки| між собою, але і з|із| частками|частинками|, що мають вищий ζ-потенціал, залучаючи останні в процес коагуляції. Роль часток, заряджених слабкіше,|частинок| і виконують гідроксиди алюмінію і заліза.
Утворення макрофази при коагуляції відрізняється від утворення такої при кристалізації солей з пересичених розчинів. Гідроксид, що коагулює, сорбується на поверхні грубодисперсних часток і одночасно утворює «клейові містки», що зв'язують ці частки між собою в комплекси (рис. 2.3). Органічні речовини (наприклад, гумінові кислоти) сорбуються на поверхні гідроокису, що коагулював. Макрофаза (флокули або пластівці), що утворюється, має вельми рихлу структуру, оскільки проміжки між частками заповнені водою. Вона має щільність (1,001 – 1,1 т/м3), що мало відрізняється від щільності води, і невисоку механічну міцність. Розмір цілком сформованих флокул складає 1 – 3 мм. Ці флокули потім виділяються з води в процесі висвітлювання.
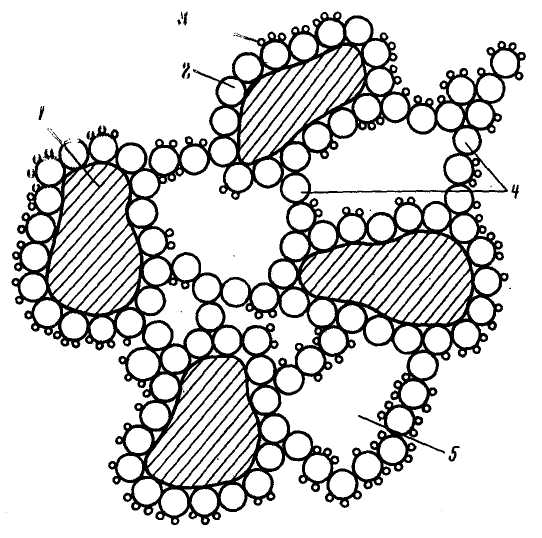
Рис. 2.3. Структура пластівців, що утворюються при коагуляції:
1 – частки ГДП; 2 – частки гідроокису; 3 – органічні речовини; 4 – «клейові» містки; 5 – «захоплена» вода.
В наш час теоретичні розробки не дають підстав для обчислення необхідної дози коагулянту для ефективного проведення процесу, тому доза підбирається експериментально. Експериментально підбирається і оптимальне необхідне значення рН. Доза коагулянту залежить від складу домішок води. Так, при великій концентрації грубодисперсних речовин і малої концентрації колоїдних домішок доза коагулянту має бути максимальна, і навпаки. Орієнтовно дозу коагулянту залежно від складу домішок води можна оцінити, користуючись таблицею 2.1.
Таблиця 2.1. Зразкова доза коагуляції залежно від складу домішок води
| Окислюваність води, мгО2/л | Концентрація ГДП, мг/л | Прозорість по «хресту», см | Доза безводного|безводної| коагулянту мг-екв/л |
| 5—8 8—12 12—15 15—20 20—25 25-30 >30 | До 50 50—100 100—200 200—400 400—600 600—800 800—1000 1000—1400 1400—1800 1800—2200 | 0,3—0,5 0,4—0,6 0,6 – 0,8 0,7—1,0 0,8—1,25 0,95—1,4 1,05—1,6 1,15–1,85 1,3—2,0 1,4—2,2 |
При вмісті в початковій|вихідній| воді грубодисперсних і колоїдних| речовин понад 100 мг/кг коагуляція проводиться у висвітлювачах, а при меншій концентрації більш економічно організувати її безпосередньо на насипних фільтрах. В цьому випадку мають справу|річ| з|із| прямотічною| або контактною коагуляцією. При контактній коагуляції процес відділення|відокремлення| пластівців відбувається у фільтруючому шарі. Контактна коагуляція представляє| особливий випадок, коли дрібні|мілкі| частки|частинки| затримуються|утримуються| на поверхні крупних зерен шару. Вона відрізняється більшою швидкістю протікання процесу і майже повним|цілковитим| видаленням|видобуванням| з|із| води дрібних|мілких| часток|частинок|.
Для інтенсифікації процесу коагуляції часто у воду, що обробляється, вводять|запроваджують| спеціальні речовини – флокулянти|. Суть|єство| процесу флокуляції полягає в тому, що агрегація колоїдних часток|частинок| в цьому випадку відбувається|походить| не лише|не тільки| безпосередньо, але і через молекули флокулянта|. В якості флокулянтов| використовуються неорганічні або органічні високомолекулярні з'єднання|сполуки|: активна кремнекислота|, поліакриламід| і ін. Так, молекула поліакриламіду| дисоціює і за кислотним, і за основним типом в залежності від рН|. У ізоелектричному стані|достатку| ступінь|міра| дисоціації поліакриламіду| за обома типами однаковий. Проте|однак| не дивлячись на|незважаючи на| наявність біля|в| молекули поліакриламіду одночасно позитивно і негативно|заперечний| заряджених іоногенних| груп в цілому|загалом| вона електронейтральна|. Іоногенні групи молекул поліакриламіду| сорбують різні частки|, утворюючи крупні структуровані системи. Слід зауважити, що флокуляція| не замінює процес коагуляції, а лише заглиблює|поглиблює| і інтенсифікує його.
При коагуляції знижується бікарбонатна лужність води. Проте еквівалентно цьому у воду при введенні коагулянту потрапляють і іони SO42–. В той же час концентрації іонів Са2+ і Mg2+ при коагуляції не змінюються і відбувається лише заміна карбонатної жорсткості на некарбонатну.
Великий вплив на процес коагуляції надає|виявляє| температура|. При підвищенні температури збільшуються швидкість і глибина гідролізу, швидкість формування і відділення|відокремлення| твердої фази, що в кінцевому|скінченному| випадку прискорює і заглиблює|поглиблює| коагуляцію домішок|нечистот|. Оптимальною для коагуляції води за сірчанокислим алюмінієм вважається температура близько 303 – 308 К.
При здійсненні технологічного процесу дуже велике значення має кінетика коагуляції. Практика показує, що для створення|створіння| оптимальних умов процесу необхідно спочатку швидке перемішування води з|із| коагулянтом (< 10 хв.|), а потім процес повинен відбуватися|походити| в спокійнішій гідродинамічній обстановці.
У загальному випадку швидкість коагуляції визначається кількістю часток|частинок|, що злипаються, в одиницю часу, причому розрізняють повільну коагуляцію, коли не кожне зіткнення |сутичка| ?????????7, і швидку, коли ????????злипанням.
???????? швидкість швидкої коагуляції ???????? описується рівнянням реакції другого порядку:
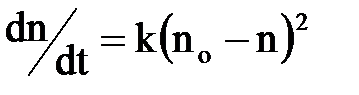
| (2.14) |
де nо – число часток на початку процесу;
n – число агрегатів, що утворилися;
k – константа коагуляції.
Представивши число часток, що залишилися, до моменту часу τ як
по – n = nτ, швидкість їх убування можна записати так:
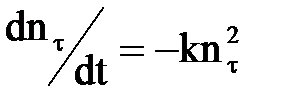
| (2.15) |
Після ділення змінних і інтегрування в межах від nо до nτ і від 0 до τ, отримаємо:
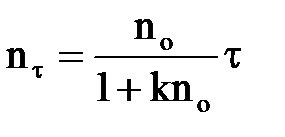 . .
| (2.16) |
Константа коагуляції визначається виразом:
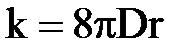 , ,
| (2.17) |
де D – коефіцієнт броунівської дифузії часток, м2/с;
r – радіус частки, м.
Число часток|частинок|, що створюють агрегати в одиниці об'єму|обсягу| і в одиницю часу, складає
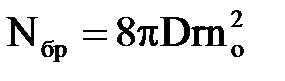
| (2.18) |
У ламінарному| потоці швидкості руху часток|частинок| відповідають епюрі| швидкостей води, тобто частки|частинки| рухаються|сунуть| з|із| різними| швидкостями. Частки|частинки|, що рухаються|сунуть| швидше, настигають повільніші, і якщо при цьому відстань між ними виявляється|опиняється| рівною сумі їх радіусів, то частки|частинки| зустрічаються| одна з|із| одною і утворюють агрегати. Для монодисперсних часток|частинок| повне|цілковите| число зустрічей визначається виразом:
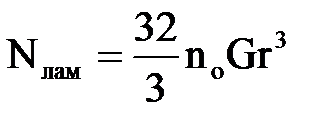
| (2.19) |
де G – швидкісний градієнт dυ/dz, с–1;
υ – відносна швидкість часток, що рухаються;
z – відстаньміж частками в плоскості, перпендикулярній швидкості руху потоку, м.
У турбулентному потоці води число часток|частинок|, що стикаються, збільшується за рахунок пульсацій потоку і складає
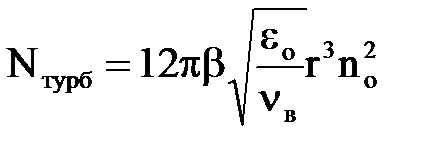
| (2.20) |
де β – експериментальний коефіцієнт;
εo – витрата енергії на пульсації потоку;
νв – кінематична в'язкість води, м2/с.
Співвідношення між ефективністю коагуляції в турбулентному потоці і броунівською коагуляцією, рівне приблизно 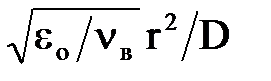 , показує, що при розмірі часток|частинок| приблизно 5 нм| мають місце обидва механізми коагуляції, для дрібніших|мілких| часток|частинок| навіть у турбулентному потоці переважає броунівська коагуляція.
, показує, що при розмірі часток|частинок| приблизно 5 нм| мають місце обидва механізми коагуляції, для дрібніших|мілких| часток|частинок| навіть у турбулентному потоці переважає броунівська коагуляція.
Режим потоку води надає|виявляє| великий вплив на формування пластівців і, з врахуванням|з урахуванням| їх неміцності, може навіть сприяти руйнуванню пластівців, що сформувалися. Тому швидкість води в зоні їх формування і відстоювання має бути не більше 1 – 1,5 мм/с.
Якщо сульфати, що вводяться|запроваджують| у воду, при коагуляції сірчанокислим| алюмінієм сприяють процесу коагуляції і частково виводяться разом з осадом, то при коагуляції сірчанокислим залізом вони практично всі потрапляють| на наступний|такий| ступінь|рівень| очищення (іонообмінну) і створюють додаткові труднощі при їх видаленні|віддаленні| з|із| води. Тому найраціональніше вводити|запроваджувати| в оброблювану воду іони цих металів без аніонів, що утрудняють процеси знесолення.
Цього можна досягти лише при застосуванні електрохімічного| методу (електрокоагуляції), заснованого на анодному| розчиненні металу у воді при проходженні через воду електричного струму|току|. При цьому очищення води від колоїдних речовин здійснюється у ряді|в ряді| процесів, що протікають одночасно: електрохімічного розчинення електродів з переходом| іонів металу в розчин, окислювально-відновлювальних реакцій на електродах, власне коагуляції, явищ електрофорезу| (руху часток|частинок| під впливом зовнішнього електричного| поля).
У основі електрокоагуляції лежить процес анодного розчинення металів під дією постійного електричного струму|току| з|із| подальшим|наступним| гідролізом катіонів металів і їх участю в процесі коагуляції домішок|нечистот| води.
Електрохімічне розчинення металів, що занурені в розчин і знаходяться|перебувають| під дією прикладеного ззовні електричного потенціалу, залежить від багатьох чинників|факторів|, але|та| в основному визначається процесами, що відбуваються|походять| на аноді і катоді.
На аноді відбувається|походить| окислення металу з|із| переходом його іонів в розчин:
| Al – 3е → Al3+; | (2.21) |
| Fe – 2е → Fe2+. | (2.22) |
Безперервний вихід іонів металу в розчин можливий лише за умови відведення електронів з катода, який здійснюється при протіканні реакцій відновлення.
????
| 2Н2О + О2 + 4е → 4ОН–. | (2.25) |
Кисень також бере участь в процесі переходу іонів Fe2+ у форму Fe3+ і при досить високому потенціалі виділяється на аноді, пасивуючи метал (утворюючи плівки оксиду). При цьому внаслідок поляризації електроду різко скорочується швидкість його розчинення. Поляризація катоду відбувається за рахунок відкладень на його поверхні лужноземельних з'єднань унаслідок високих значень рН води в прикатодній області.
Іони ОН– утворюють з іонами металу гідроокису: Аl(ОН)3 або Fe(ОН)2. Гідрозакис заліза потім окислюється в гідроокис:
| 4Fe(ОН)2 + О2 + Н2О → 4Fe(ОН)3. | (2.26) |
Таким чином, гідроокиси металів можуть бути отримані у воді без введення в неї аніонів SO42–. Крім того, електрохімічний метод електрокоагуляція дозволяє відмовитися від використання традиційних коагуляцій, а також апаратури, пов'язаної з їх приготуванням і дозуванням.
Теоретично кількість електрики, необхідної для розчинення 1 г-екв металу, складає 26,8А·r. Проте практично кількість електрики завжди вище теоретичної внаслідок поляризаційних ефектів на пластинах, витрати енергії на нагрів води і т.п. Для зменшення цього ефекту і більш рівномірного використання пластин проводять через певний час (зазвичай через 15 мін) переполюсовку напруги, що підводиться. При проведенні електрокоагуляції спостерігається підвищення рН води на 0,5 – 1,0 за рахунок розрядки іонів Н+, що дає можливість не підлужувати воду реагентами.
Практично у всіх апаратах для електрокоагуляції, що застосовуються в промисловості, використовуються пластини металу, що підвищує вартість обробки і збільшує габарити апарату (рис. 2.4). Всі конструкції пластинчастих апаратів відносяться до безнапірного типу. При роботі з великими витратами води такі апарати незручні, їх складно розташовувати в схемах ВПУ. Вживання пластинчастих апаратів невигідне також і тому, що використання пластин можливе тільки до 50%-го їх розчинення. Інакше внаслідок викривлення пластин можливе коротке замикання в апараті.
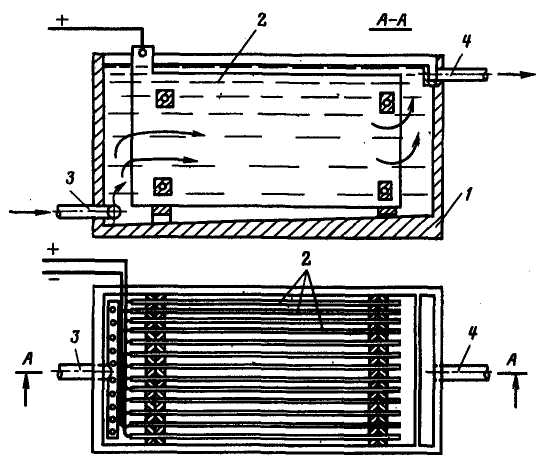
Рис. 2.4. Пластинчаста електрокоагуляція:
1 – корпус; 2 – пластини з металу; 3 – надходження води; 4 – вихід води.
Існує два способи підключення електродів до джерела живлення|харчування|: монополярний, коли однойменні пластини (наприклад, аноди) підключаються до одного з полюсів джерела живлення|харчування|, а катоди – до іншого, і біполярний, коли живлення|харчування| підводиться лише до крайніх електродів і спільне|загальне| падіння напруги|напруження| на апараті складається з|із| суми падінь напруги|напруження| на окремих ячейках|комірках|.
При біполярному підключенні електродів потрібна більша| напруга|напруження| на апарат, але|та| менша кількість контактів. При цьому зменшуються розміри апарату і перетин кабелів, що підводяться, що спрощує монтаж і експлуатацію електрокоагуляторів| і зменшує їх вартість.
Напруга, що рекомендується, на ячейках при відстані між електродами 10 – 12 мм для анодного розчинення заліза складає 3 В, а для розчинення алюмінію – 4 В. Із досвіду освоєння електрокоагуляторів рекомендується підтримувати щільність струму близько 10 А/м2, а швидкість потоку води між пластинами – не менше 0,5 м/с. Витрата електроенергії на електрокоагуляцію складає 0,05 – 0,5 кВт · ч/т води.
Перспективні апарати з використанням в якості анодів масивних металевих відливок (рис. 2.5). Така електрокоагуляція складається з масивного аноду, встановленого в баку з неелектропровідного матеріалу і
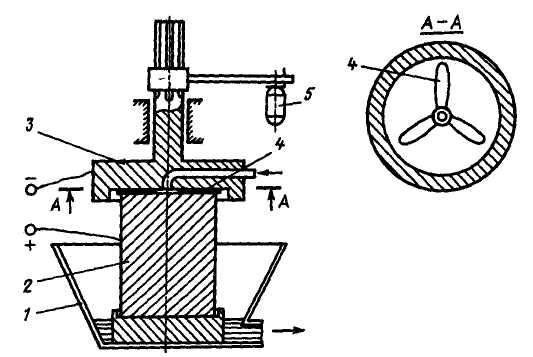
Рис. 2.5. Електрокоагуляція з поверхнею аноду, що оновлюється:
1 – збірна ванна; 2 – розчинний анод; 3 – катод; 4 – пластинки з абразивного матеріалу; 5 – сервопривід; → – очищена вода
розташованого над ним на відстані 0,5 – 1 мм катода. Малий зазор стабілізується за допомогою пластинок з абразивного матеріалу, які при обертанні катоду видаляють з аноду пасивуючу плівку оксиду металу. Вода подається через центральний отвір в аноді (або через катод) і протікає в щілині між катодом і анодом зі швидкістю, що забезпечує турбулентний режим потоку. Мала величина зазору дозволяє різко скоротити втрати енергії на електричний опір води і підвищити щільність струму більш ніж у 100 разів в порівнянні з пластинчастими електрокоагуляторами. Катоду може бути надано або поворотно-обертальний рух за допомогою кулісного механізму, або обертальний з використанням енергії потоку води. Така конструкція автоматично забезпечує постійну величину зазору при спрацьовуванні анода.
Підвищити інтенсивність процесу анодного розчинення металу можна при збільшенні площі електродів. Цього можна добитися, не збільшуючи габарити апаратів, за допомогою засипних електродів з|із| гранул металів. Крім усього іншого цей метод дозволяє також використовувати відходи металу і понизити|знизити| вартість обробки води.
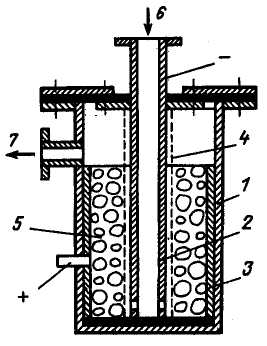
Рис. 2.6. Електрокоагуляція типу «труба в трубі» з пористою засипкою:
1 – корпус; 2 – внутрішня труба-катод; 3 – вставний анод; 4 – сітка; 5 – гранули металу; 6 – вхід води; 7 – вихід води.
Конструкція такого апарату типу «труба в трубі», розробленого в МЕІ, представлена на рис. 2.6. У цьому апараті внутрішня труба використовується для подачі води і одночасно служить катодом. Анодом є труба більшого діаметру, вставлена в корпус і прилегла до нього зсередини. Пористе середовище розташовується в міжтрубному просторі і відділяється від катода перфорованою неелектропровідною прокладкою. Розташування основних електродів дозволяє розподілити прикладений потенціал таким чином, що щільність струму на основному аноді буде мінімальною, що дає можливість продовжити час його роботи. Подача води знизу до верху сприяє евакуації газу з порожнини апарату. Розташування шару дозволяє йому у міру спрацьовування під силою тяжіння опускатися вниз, рівно заповнюючи простір між анодом і катодом. Ця конструкція може бути виконана і в біполярному варіанті підключення електродів, якщо встановити кільцеподібні перфоровані діафрагми, що розділяють пористе середовище на окремі шари. Такий апарат може працювати під тиском, що істотно розширює можливість його вживання в схемах ВПУ ТЕС.
Ефективність електрокоагуляційного очищення води істотно вище за ефективність реагентної| коагуляції і складає 70 – 90% проти|супроти| 50 – 60% при реагентній| коагуляції.
У загальному вигляді|виді| розрахунок концентрації розчинених іонів в електрокоагуляторі типу|типу| «труба в трубі» зводиться до встановлення взаємозв'язку між гідродинамічними|, електрохімічними і електричними параметрами апарату.
Швидкість анодного розчинення металу рівна
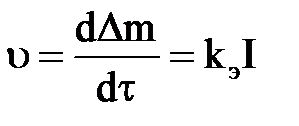 , ,
| (2.27) |
де Δm – маса розчиненого металу, г;
I – сила струму, А;
kе – електрохімічний еквівалент [наприклад, для алюмінію kе = 0,336 г/(А· ч)].
Тоді теоретична концентрація іонів металу у воді визначається як
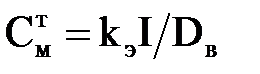 , ,
| (2.28) |
де Dв – витрата води, м3/ч.

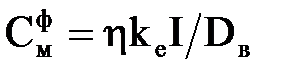 , ,
| (2.30) |
В той же час необхідна для виділення такої кількості іонів ???????? сила струму в апараті визначається як:
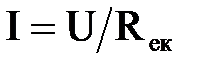 ,
,
де U – напруга постійного струму;
Rек – спільний електричний опір електрокоагулятора.
Підставляючи значення I у (2.30), отримуємо
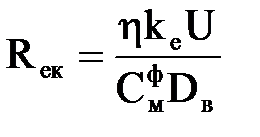 , ,
| (2.31) |
а з урахуванням того, що Dв = υвSек,
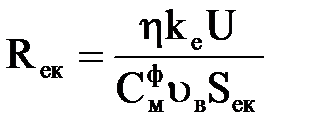 , ,
| (2.32) |
де υв – швидкість води в порожнині електрокоагулятора, м/ч;
Sек – площа поперечного перетину апарату, м2.
У апараті типу «труба в трубі» за рахунок спрацьовування гранул і в результаті їх опускання в порожнині апарату утворюється вільний простір, заповнений водою. Тоді спільний електричний опір Rек,може бути представлено в наступному вигляді:
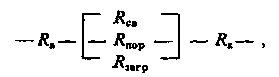
де Rа і Rк – відповідно електричний опір на основному аноді і катоді унаслідок поляризаційних ефектів;
Rсв – електричний опір вільного об'єму, заповненого водою;
Rпор – електричний опір об'єму води в порах завантаження;
Rзагр – електричний опір завантаження.
Електричний опір об'єму|обсягу| води між коаксіальними циліндричними| електродами
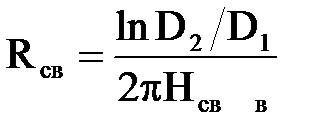
| (2.33) |
де D2 і D1 – відповідно діаметри зовнішнього і внутрішнього електродів, см;
Нсв –висота електродів над завантаженням, см;
Електропровідність води ϰв може бути визначена експериментально або обчислена з вираження
ϰв 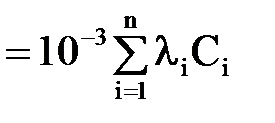
| (2.34) |
де Сі – концентрація i-го іону, г-екв/л;
λi – еквівалентна електропровідність, См·см2/г-екв.
Дослідні дані показують, що значення Rсв, визначене експериментально, вище, ніж Rсв, обчислене по (2.33) і (2.34). Це відбувається через наявність поляризаційних ефектів на основних електродах. Тому надалі в розрахунок повинне прийматися значення величини ϰв з формули, аналогічній формулі (2.33), де Rсв визначається експериментально.
Електричний опір води в порах завантаження|загрузки| визначається як
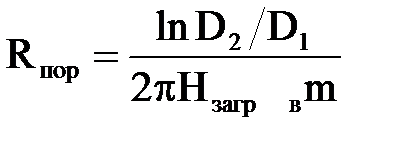
| (2.35) |
де m– порозность завантаження, що залежить від форми гранул металу і їх укладання в завантаженні;
Нзагр – висота завантаження.
Загальний електричний опір електрокоагулятора з врахуванням|з урахуванням| поляризаційних| ефектів на основних електродах по другому закону Кирхгофа рівний
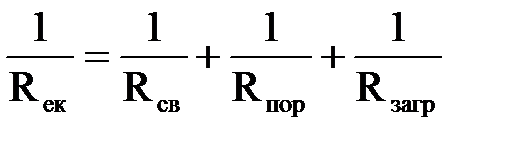 , ,
| (2.36) |
звідки можна в явному вигляді|виді| визначити
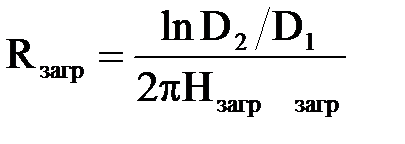 , ,
| (2.37) |
де ϰзагр – величина, що характеризує електропровідність завантаження.
Підставивши в (2.36) значення Rек, Rсв, Rпор, Rзагр і провівши спрощення, отримаємо вираження, що зв'язує електричні, електрохімічні і конструктивні характеристики електрокоагулятора, яке з урахуванням того, що
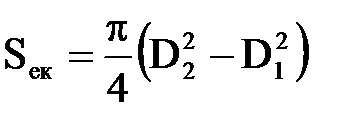
| (2.38) |
| ϰ |
| ϰ |
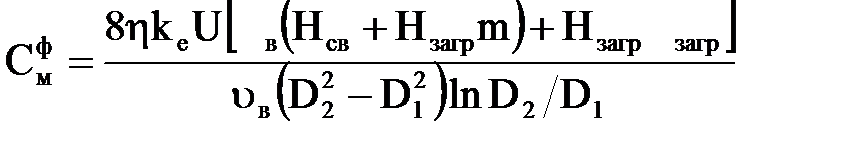
| (2.39) |
Використання цього рівняння вимагає попереднього експериментального визначення в лабораторних умовах для реальної води значень Rек, Rсв і т при різних відношеннях D2/D1 і швидкостях потоку води.
Дата добавления: 2015-05-19; просмотров: 1948;