Генетически обусловленные дефекты биосинтеза гормонов
Биосинтез любого гормона представляет собой сложный многозвеньевой процесс, в котором принимает участие множество ферментов. При этом образование любого фермента, точнее, его апофермента, определяется активностью соответствующего гена. Мутация гена может привести к недостаточности образования апофермента или такому его изменению, при котором образующийся фермент теряет свою активность. В этом случае нарушается последовательный ход биосинтеза соответствующего гормона, что обусловливает: 1) гипофункцию железы; 2) накопление в железе промежуточных продуктов биосинтеза, образующиеся до места блокады, которые выделяются в кровь и оказывают специфический патофизиологический эффект; 3) нарушение механизма обратной связи и развитие дополнительных патологических процессов. Иллюстрацией к этому служат два примера.
Первый пример. На рис. 20-3 в общих чертах представлен биосинтез кортизола и участки его блокады. В настоящее время хорошо изучены два вида блокады образования кортизола в связи с дефицитом ферментов - 21-гидроксилазы (I) в одном случае и 11 β-гидроксилазы (II) - в другом. При дефиците 21-гидроксилазы (I) процесс биосинтеза заканчивается образованием прогестерона и 17а-оксипрогестерона. Кортизол не образуется. Это по механизму обратной связи растормаживает секрецию кортиколиберина в гипоталамусе, что, в свою очередь, ведет к усилению образования АКТГ. АКТГ стимулирует стероидогенез до места блокады, и так как кортизол не образуется, то вся эта стимуляция переключается на образование D4-андростен-3,17-диона, обладающего андроген-
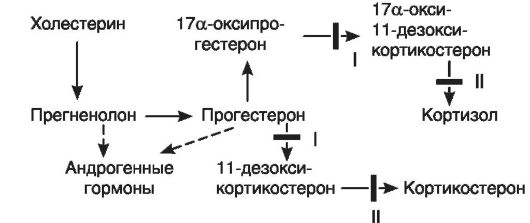 Рис. 20-3.Участки блокады биосинтеза кортизола
Рис. 20-3.Участки блокады биосинтеза кортизола
ными свойствами. Его поступление в кровь значительно увеличивается. Образующиеся в надпочечниках андрогены включаются в механизм обратной связи, регулирующей развитие половых желез, и приводят к выключению этой регуляции, что сопровождается атрофией половых желез как у мальчиков, так и у девочек. Дефект выявляется уже в период эмбрионального развития. У эмбриона женского пола к этому периоду внутренние половые органы уже заложены, поэтому избыток андрогенов вызывает их гипоплазию и развитие вирилизма. Маскулинизация продолжается и после рождения. У мальчиков же появляются признаки преждевременного полового созревания.
Подобный механизм включается и при дефекте фермента 11b-гидроксилазы (II). Кортизол также не образуется, но в этом случае (в отличие от предыдущего синдрома) накапливается избыточное количество 11-дезоксикортикостерона и 17а-окси-11- дезоксикортикостерона, первый из которых обладает выраженными минералокортикоидными свойствами. Это ведет к повышению кровяного давления. Всю эту патогенетическую цепь можно разорвать введением глюкокортикоидов (рис. 20-4). Они тормозят образование АКТГ и тем самым уменьшают образование андрогенов.
Второй пример. Биосинтез тиреоидных гормонов, происходящий в клетках фолликулярного эпителия щитовидной железы,
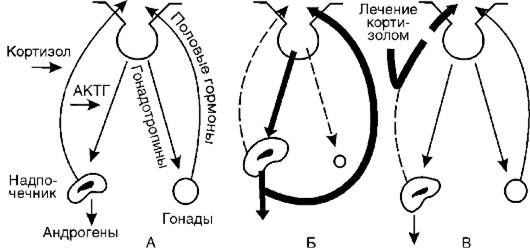 Рис. 20-4.Механизм атрофии половых желез при врожденном адреногенитальном синдроме и механизм лечебного действия кортизола: А - регуляторные механизмы в норме; Б - адреногенитальный синдром; В - патогенетическая терапия кортизолом (по Гоффу); АКТГ - адренокортикотропный гормон
Рис. 20-4.Механизм атрофии половых желез при врожденном адреногенитальном синдроме и механизм лечебного действия кортизола: А - регуляторные механизмы в норме; Б - адреногенитальный синдром; В - патогенетическая терапия кортизолом (по Гоффу); АКТГ - адренокортикотропный гормон
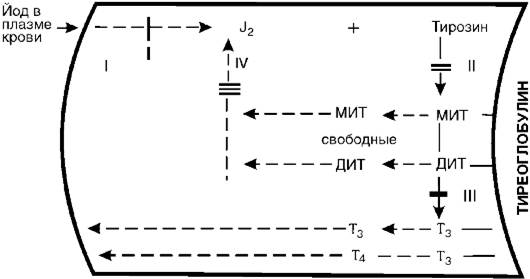 Рис. 20-5.Участки блокады биосинтеза тиреоидных гормонов. МИТ - монойодтирозин, ДИТ - дийодтирозин
Рис. 20-5.Участки блокады биосинтеза тиреоидных гормонов. МИТ - монойодтирозин, ДИТ - дийодтирозин
также является сложным многозвеньевым процессом. В общих чертах он представлен на рис. 20-5 и состоит из следующих основных процессов: 1) захват йода железой и окисление его пероксидазой в молекулярный йод или йодит; 2) йодирование тирозина тирозинйодиназой с образованием монойодтирозина (МИТ) и дийодтирозина (ДИТ); тирозин, как и МИТ и ДИТ, находится в составе тиреоглобулина; 3) конденсация молекул МИТ и ДИТ с образованием трийодтиронина (Т3) и тироксина (Т4); 4) образование свободных МИТ и ДИТ и их дегалогенизация; выделяющийся при этом йод снова идет на йодирование тирозина. В связи с дефектами соответствующих ферментов каждый из указанных этапов может блокироваться.
Установлена возможность блокады йодзахватывающей системы (I). Для этого случая характерна неспособность железы поглощать J131 при соответствующем исследовании. Исправление этого дефекта достигается введением в организм небольших доз йодистого калия, который в связи с повышением его концентрации в крови, в силу диффузии проникает в щитовидную железу и, таким образом, компенсирует дефект йодзахватывающей системы. II - блокада йодирования тирозина. Поглощенный йод сохраняется в железе в неорганической форме и не включается в тирозин. Этот дефект на данном этапе компенсируется введением готовых тиреоидных гормонов. III - дефект конденсации йодтирозинов. Характеризуется
накоплением промежуточных продуктов - МИТ и ДИТ и следовыми количествами Т3 и Т4. Компенсация дефекта проводится также введением гормонов. IV - дефект йодтирозин-дегалогеназы. Он характеризуется угнетением дегалогенизации МИТ и ДИТ. Эти продукты накапливаются, выделяются в кровь и выводятся из организма. Организм теряет йод, развивается йодная недостаточность. Компенсация дефекта может быть обеспечена введением в организм йодистого калия.
Каждый из указанных дефектов приводит к недостаточному образованию тиреоидных гормонов. В результате возникает гипофункция щитовидной железы, сопровождаемая развитием зоба (увеличением щитовидной железы) и кретинизма. Последнее объясняется тем, что эти дефекты возникают еще до рождения или в детском возрасте.
Дата добавления: 2015-01-29; просмотров: 1993;