АДСОРБЦИЯ НА ГРАНИЦЕ ТВЕРДОЕ ТЕЛО — ЖИДКОСТЬ (АДСОРБЦИЯ ИЗ РАСТВОРОВ)
В случае адсорбции из растворов, в отличие от чистых жидкостей, жидкая фаза состоит из двух или нескольких компонентов. Это имеет огромное практическое значение, главным образом, в связи с проблемой очистки жидкостей. Адсорбция, используемая в практике человечества в течение многих веков, остается и в настоящее время, в эпоху небывалого технического прогресса, основным методом извлечения примесей и очистки жидкостей. В связи с нарастающим значением проблемы удаления промышленных сбросов и регенерации природных вод исследования адсорбции из растворов приобретают особое значение. К этому же типу адсорбции можно отнести адсорбцию ПАВ на твердом адсорбенте.
Явления адсорбции из растворов классифицируются прежде всего по адсорбату. Различают адсорбцию нейтральных молекул (неэлектролитов), ионов (электролитов) и коллоидных частиц.
5.1 АДСОРБЦИЯ НЕЭЛЕКТРОЛИТОВ (МОЛЕКУЛЯРНАЯ АДСОРБЦИЯ)
Адсорбция из растворов — процесс более сложный, чем адсорбция газов, прежде всего потому, что наряду с силовым полем твердой фазы необходимо учитывать межмолекулярные взаимодействия в жидкой фазе, которые во многих случаях становятся доминирующими. Дальнейшее усложнение системы связано с тем, что из жидкой фазы адсорбируются по меньшей мере два компонента — растворитель (1) и растворенное вещество (2) (одно или несколько) — и конкуренция между ними за места в поверхностном слое приводит к тому, что поверхностные избытки компонентов Xi оказываются различными по знаку. Если большинство мест занято молекулами растворенного вещества, происходит вытеснение растворителя из поверхностного слоя (X2>0, X1<0), чего не наблюдается, как правило, при адсорбции газов. Избытки определяют экспериментально по изменению концентрации компонента в растворе в процессе адсорбции (по анализу состава жидкой фазы до и после адсорбции),.
Избыток Xi (в молях на единицу массы адсорбента) может быть выражен по Гиббсу (при постоянном числе молей растворителя) как
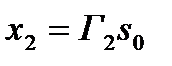 .
.
Однако при исследовании адсорбции из растворов используют обычно другие способы выражения адсорбции, например, избыток выражают через молярные объемные концентрации до (Сi0) и после (Ci) адсорбции:
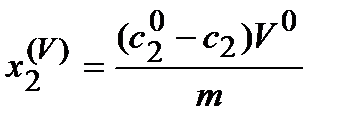 ,
,
где V0 - общий объем раствора.
Явление адсорбции из растворов широко используется для разделения многокомпонентных систем. Этот метод анализа и разделения, называемый хроматографией, был разработан русским ученым М.С.Цветом в начале XX в. Пропуская раствор хлорофилла через колонку с адсорбентом (окисью алюминия), Цвет установил, что различные компоненты сложного раствора адсорбируются на разных уровнях высоты колонки. После нескольких циклов промывания растворителем в колонке обнаруживаются расположенные одна над другой резко очерченные (по-разному окрашенные) зоны. Очевидно, что верхняя зона будет занята компонентом, обладающим наибольшей адсорбционной способностью; последующие зоны располагаются сверху вниз в порядке уменьшения адсорбционной способности. Разрезая колонку по зонам (в том случае, если они окрашены) и затем десорбируя, можно препаративно разделить компоненты.
Наряду с адсорбционной существуют и другие виды хроматографии: ионообменная, распределительная, осадочная. Основные способы анализа — фронтальный, элюентный и вытеснительный. Все они рассматриваются в специальных курсах.
Таким образом, молекулярная адсорбция из растворов широко используется для очистки жидкостей, извлечения ценных примесей, оценки удельной поверхности, а также для разделения и анализа многокомпонентных систем.
5.2 АДСОРБЦИЯ ЭЛЕКТРОЛИТОВ
Ионы в растворе являются носителями электрического заряда. Поэтому адсорбция ионов (переход из объемной фазы в поверхностный слой) сопровождается перераспределением зарядов, возникновением электрического поля в области поверхностного слоя. Например, переход катионов из объемной жидкой фазы на границу с твердой поверхностью приводит к тому, что последняя заряжается положительно, жидкая - отрицательно и в поверхностном слое возникает двойной электрический слой (ДЭС) зарядов, подобный плоскому конденсатору с двумя заряженными обкладками. ДЭС возникает при любом направлении перехода ионов одного знака. Поскольку адсорбция ионов неразрывно связана с образованием ДЭС, необходимо вначале познакомиться более детально с происхождением двойного электрического слоя и его свойствами.
5.2.1 ДВОЙНОЙ ЭЛЕКТРИЧЕСКИЙ СЛОЙ
Возможны два принципиально разных пути образования ДЭС: избирательная адсорбция поверхностью твердой частицы ионов из дисперсионной среды и ионизация поверхностных молекул твердой частицы.
При избирательной адсорбции возможны два варианта.
а) Избирательная адсорбция ионов, способных достраивать кристаллическую решетку частицы дисперсной фазы.
В соответствии с правилом Панета-Фаянса, на поверхности твердой частицы избирательно адсорбируются только те ионы, которые способны достроить ее кристаллическую решетку или изоморфны с ней. Предположим, что твердые частицы хлорида серебра диспергированы в водном растворе хлорида калия. В данном случае на поверхности будут сорбироваться ионы С1-, так как они входят в состав кристаллической решетки и будут придавать частице отрицательный заряд, одновременно прилегающая к частице жидкая среда приобретает положительный заряд, т. е. возникает ДЭС. Ионы, которые придают заряд твердой частице, называются потенциалопределяющими, противоположно заряженные - противоионами.
б) Избирательная адсорбция без достройки кристаллической решетки. Этот вариант имеет место, когда в растворе имеются ионы, обладающие большой адсорбционной способностью - Н+ или ОН-.
При ионизация поверхностных молекул твердой частицы, также возможны два случая.
а) В случае гидрозолей металлов в раствор переходят катионы металлов, твердая поверхность заряжается отрицательно, а дисперсионная среда положительно.
б) В случае некоторых оксидов, кислот, белков и т. д. с твердой поверхности в дисперсионную среду переходят ионы одного заряда, ионы с противоположным зарядом остаются на твердой частице и являются потенциалопределяющими.
Существует несколько теорий строения ДЭС, отметим наиболее значительные:
• теория Гельмгольца-Перрена (1879);
• теория Гуи-Чэпмена (1910-1913);
• теория Штерна (1924).
Отличие между этими теориями сводится, в основном, к различному толкованию структуры слоя противоионов. Современные представления строения ДЭС:
1. ДЭС образован потенциалопределяющими ионами, находящимися на поверхности твердой частицы и эквивалентным количеством противоионов, находящихся в дисперсионной среде вблизи поверхности твердой частицы.
2. Потенциалопределяющие ионы прочно связаны с твердой частицей хемосорбционными силами и равномерно распределены по ее поверхности.
3. Дисперсионная среда рассматривается как непрерывная (бесструктурная) среда, характеризуемая диэлекрической проницаемостью ε и вязкостью η.
4. ДЭС рассматривается как плоскопараллельный. Это допущение приемлемо, так как толщина ДЭС намного меньше, чем радиус кривизны поверхности твердой частицы.
5. Противоионы имеют конечные размеры и, следовательно, не могут подходить к твердой поверхности ближе, чем на расстояние одного ионного радиуса.
6. Слой противоионов, компенсирующих заряд твердой поверхности, имеет сложное строение и состоит из двух частей: плотного слоя (адсорбционного слоя, или слоя Гельмгольца) и диффузного, т.е слоя Гуи.
7. Адсорбционный слой противоионов примыкает к заряженной поверхности твердой частицы и имеет толщину порядка диаметра гидратированного противоиона d. Те противоионы, которые находятся в этом пространстве, называются адсорбционными противоионами. Они связаны с заряженной твердой частицей двумя видами сил -адсорбционными и электростатическими. Эта связь является настолько прочной, что противоионы адсорбционного слоя перемещаются вместе с твердой частицей, не отрываются от нее, образуя с ней единое кинетическое целое — коллоидную частицу. Противоионы адсорбционного слоя равномерно распределены в слое, поэтому падение потенциала (ψd) происходит линейно.
8. Диффузный слой имеет толщину δ, его образуют те противоионы, которые находятся от заряженной поверхности на расстоянии, большем d, но в пределах расстояния δ. Эти противоионы притягиваются к частице только электростатическими силами, следовательно, менее прочно, чем противоионы адсорбционного слоя. При движении твердой частицы они от нее отрываются. На противоионы диффузного слоя большое влияние оказывает тепловое движение, которое стремится распределить их равномерно по всему объему системы. Его действие тем сильнее, чем дальше от заряженной поверхности находятся противоионы. Это приводит к установлению динамического равновесия в диффузном слое. Так как противоионы в диффузном слое распределены неравномерно, то и падение потенциала (ψδ) в нем происходит также неравномерно - по криволинейной зависимости.
9. Полное падение потенциала в ДЭС называется термодинамическим потенциалом ψ0:
ψ0 = ψd + ψδ .
В ДЭС происходит полная компенсация суммарного заряда твердой поверхности суммарным зарядом противоионов и на границе ДЭС с дисперсионной средой потенциал равен нулю. Схема строения ДЭС представлена на рис. 32.
Электрически нейтральную коллоидную частицу называют мицеллой. Строение мицеллы лиофобного золя AgCl можно представить в виде формулы:
{n[AgCl]mAg+(m-x)NO3-}x+ · xNO3-,
где п - число молекул, образующих дисперсную частицу; т - число потенциалопределяющих ионов; (т-х) — число противоионов адсорбционного слоя; х - число противоионов диффузного слоя.
При движении частицы двойной электрический слой разрывается. Меcто разрыва при перемещении твердой и жидкой фаз относительно друг друга называется плоскостью скольжения или границей скольжения. На рис. 32 плоскость скольжения обозначена линией АВ. Плоскость скольжения лежит или на границе между диффузным и адсорбционным слоем, или в диффузном слое, но вблизи этой границы.
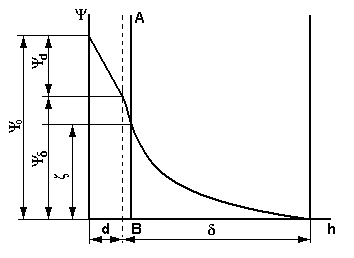
|
| Рис. 32. Строение двойного электрического слоя |
Потенциал на плоскости скольжения называется электрокинетическим потенциалом, или дзета-потенциалом ( ζ ). Дзета-потенциал является важнейшей характеристикой ДЭС: он определяет скорость относительного перемещения дисперсной фазы и дисперсионной среды, интенсивность электрокинетических явлений, устойчивость золей и т.д.
Величина дзета-потенциала определяется:
• величиной термодинамического потенциала ψ0 и характером падения потенциала в ДЭС;
• характером движения жидкости вблизи твердой поверхности (он определяет местонахождение плоскости скольжения), который зависит главным образом от вязкости среды.
Дзета-потенциал тем больше, чем больше термодинамический потенциал ψ0.
Дзета-потенциал тем больше, чем больше δ, т.е, чем больше противоионов находится в диффузном слое (при одном и том же ψ0).
Введение в золь растворов электролитов приводит к изменению строения ДЭС и, как следствие, значения дзета-потенциала.
В зависимости от природы и концентрации электролита может происходить:
• сжатие ДЭС и уменьшение абсолютной величины дзета-потенциала;
• увеличение абсолютных величин термодинамического и дзета-потенциала;
• перезарядка поверхности частицы.
Значение дзета-потенциала зависит также от величины рН, температуры и природы дисперсионной среды.
Поверхность твердых частиц, находящихся в жидкой дисперсионной среде, приобретает электрический заряд в результате преимущественной адсорбции одного из ионов электролита либо диссоциации поверхностных ионогенных групп. Независимо от механизма возникновения заряда на коллоидной частице возникает двойной электрический слой (ДЭС), состоящий из ионов на поверхности (потенциалопределяющих ионов) и из компенсирующих заряд поверхности ионов (противоионов) в растворе; причем часть противоионов находится в прилегающем к поверхности и прочно связанном с ней адсорбционном слое, а другая часть - в диффузном слое, удаленном от поверхности.
Вслед за образованием ДЭС в начальный момент контакта двух фаз все последующие процессы, происходящие в ДЭС, представляют, в основном, обмен ионов во внешней обкладке - вторичный адсорбционный процесс. Поэтому адсорбцию электролитов во всех последующих стадиях можно рассматривать, как эквивалентный обмен противоионов.
5.2.2 ИОННЫЙ ОБМЕН
Ввиду огромного значения ионного обмена для теории и практики, следует подробнее познакомиться с его основными закономерностями.
Прежде всего, отметим отличия от молекулярной адсорбции. Так, уже при первичной адсорбции электролита, где из раствора в поверхностный слой формально переходят молекулы в целом, в действительности заряженные ее «части» — ионы — располагаются неодинаково, образуя внутреннюю и внешнюю обкладки.
Вторичный процесс — ионный обмен — отличается тем, что из раствора в ДЭС уходит лишь один из ионов, и этот процесс всегда сопровождается десорбцией эквивалентного количества других ионов в раствор.
Практически обмен ионов идет на любой твердой поверхности, находящейся в растворе электролита, поскольку все твердые тела в той или иной степени оказываются гетерополярными. Так, типично неполярный адсорбент — уголь — при взаимодействии с кислородом воздуха или воды образует поверхностные (хемосорбционные) соединения — оксиды различного типа.
Такие неполярные вещества, как парафин, полиэтилен, фторопласт и другие также приобретают в растворах поверхностный заряд за счет образования оксидных групп (обычно по местам дефектов структуры) или преимущественной первичной адсорбции Н+, ОН- или других ионов из раствора.
Рассмотрим ионообменные смолы, широко используемые на практике. Эти вещества состоят из жесткой высокомолекулярной матрицы (каркаса, сетки, скелета), включающей фиксированные ионы одного знака и пропитанной раствором, который содержит в основном подвижные противоионы. Такие студни, изображенные на рис. 33, получаются, например, окислением полимерной сетки с образованием карбоксильных групп или сульфирования (обработкой концентрированной H2S04 или олеумом), вводящего сульфогруппы в матрицу. При контакте с водными растворами эти вещества диссоциируют на анионы, закрепленные в матрице, и ионы Н+, выходящие в раствор, пропитывающий матрицу, по типу: R(SО3-)n | nН+, где R — матрица. Такие системы, способные к обмену подвижных Н+ на катионы раствора, называются катионитами (рис. 33).
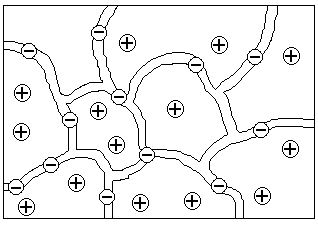
|
Рис. 33. Модель матрицы полиэлектролита (катионита) с фиксированными анионами и подвижными противоионами:
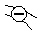 - матрица с фиксированными ионами; - матрица с фиксированными ионами;
 - противоионы - противоионы
|
В матрицу ВМС могут быть введены и группы с основными свойствами, например амино- или пиридиниевые группы, для которых характерна диссоциация по типу: R'(NН3)m | mОН-. В этом случае матрица приобретает положительный заряд (за счет фиксированных NН3-групп) и такие смолы, осуществляющие анионный обмен, называются анионитами.
Рассмотренные ионообменные смолы, широко используемые на практике для поглощения катионов или анионов из растворов, обычно называют ионитами. Этому термину следует придать более общий смысл и называть ионитами все вещества и материалы, способные к ионному обмену в растворе, подразделяя их на высокодисперсные (гетерогенные ионные адсорбенты) и высокомолекулярные иониты (ионообменные смолы); и те, и другие, в свою очередь, делятся на аниониты и катиониты.
При описании явлений обмена, общих для обоих классов, следует пользоваться термином "сорбция" (поглощение) вместо "адсорбции" - понятия, связанного с поверхностью раздела.
Системы фиксированные ионы — подвижные противоионы целесообразно называть обменным комплексом, объединяя этим термином различные способы фиксации ионов в гомогенной матрице или же во внутренней обкладке ДЭС на границе раздела фаз. Введение таких обобщающих терминов считается целесообразным для отражения той глубокой общности, которая существует между суспензоидами и молекулярными коллоидами и проявляется в ионном обмене (как и в других явлениях) в единстве основных закономерностей.
Эти основные закономерности, общие для обоих классов ионитов, устанавливались исторически в работах почвоведов уже в начале XX в., понимавших огромную роль ионного обмена в агрохимии. К.К.Гедройц в своих фундаментальных работах установил, что на почвах и грунтах происходит обмен катионов в строго эквивалентных количествах; катионы различаются по своей адсорбционной способности — способности вытеснять (в эквивалентном количестве) противоионы из поверхностного слоя на границе частиц почвы с почвенным раствором. На основании многочисленных экспериментов он расположил катионы по адсорбционной способности в следующий ряд:
Аl3+ > Ва2+ > Са2+ > Мg2+ > К+ > NН4 + > Nа+ > Li+
Видно, что адсорбционная способность увеличивается с возрастанием заряда иона, а для равнозарядных ионов - с уменьшением радиуса гидратированного иона.
Представленный ряд, называемый лиотропным, характерен и для многих других, не только гетерогенных (высокодисперсных), но и для гомогенных (высокомолекулярных) ионитов.
Ион Н+ не занимает определенного места в ряду. Для почв, грунтов и многих других объектов он стоит перед А13+, тогда как для других ионитов он располагается в конце ряда. Эти особые свойства Н+ связаны со степенью диссоциации кислот, образующих фиксированные анионы. В почвах, грунтах (а также в белковых и многих других объектах) обменный комплекс образуется в результате диссоциации слабых кислот (поликремниевых, гуминовых), характеризующихся прочной связью кислотного остатка с Н+ (водородной связью). В то же время соли этих кислот обычно хорошо диссоциированы. Поэтому Н+ вытесняет легко все остальные катионы из внешней обкладки и в почвах (при рН=6,5) занимает около половины мест в обменном комплексе. Такая же прочная связь присуща и слабокислотным (карбоксильным) высокомолекулярным ионитам, тогда как для ионитов сильнокислотного типа (с фиксированными ионами, образованными сильными кислотами, например, RSO3H) H+ не обладает высокой энергией связи и расположен в конце ряда среди одновалентных катионов.
Подобный же ряд анионов с особыми свойствами ОН- имеет место и для анионитов. Для слабоосновных ионитов, ОН- стоит в начале ряда (перед SO42- , С2О42- и других), тогда как для сильноосновных (например, образованных четвертичными аммониевыми основаниями) — в конце.
Как для теории ионного обмена, так и для всех практических применений, важно прежде всего знать, какое количество иона может быть поглощено из раствора единицей массы ионита в данных условиях, т. е. получить уравнение ионного обмена, соответствующее изотерме адсорбции. Очевидно, что в условиях конкуренции двух или более ионов одного знака за место (в водных растворах солей всегда присутствуют Н+ и ОН- и число компонентов одного знака ≥ 2), ион данного вида будет поглощаться тем больше, чем сильнее его сорбционная способность и выше его активность в растворе.
В литературе приводится целый ряд уравнений изотермы ионного обмена. Наиболее строгое решение на основе термодинамического анализа, сходного с выводом уравнения закона действия масс было получено Б.П. Никольским:
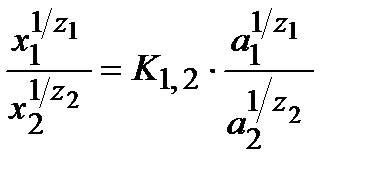 , (40)
, (40)
где Xi — поглощенное количество; zi — заряд ионов 1 и 2 с одинаковым знаком заряда; К1,2 — константа обмена этих ионов; аi— активность в равновесном растворе.
Константа К1,2 имеет физический смысл константы равновесия процесса ионного обмена и является количественным выражением отношения сорбционных способностей обоих ионов. Действительно, если в растворе а1 = а2 = 1, то К1,2 равна отношению поглощенных количеств (в степенях 1/zi;) этих ионов.
Уравнение (40) —фундаментальное соотношение, справедливое как для высокомолекулярных, так и для высокодисперсных ионитов и независимое от механизма (химического или адсорбционного) процесса. Эта общность процессов и систем согласуется с положениями, высказанными ранее. Уравнение Никольского получило экспериментальное подтверждение и широко используется в практике ионного обмена. Для разбавленных растворов вместо аi можно пользоваться равновесными значениями концентраций, определяемых аналитически.
Если соотношение поглощенных количеств двух (или нескольких) ионов определяется значениями K.ij и аi то общее количество поглощенных ионов зависит от поглощающей способности.
Поглощающую способность ионита определяют как число моль-экв ионов, поглощенных 1 кг ионита (или ммоль-экв/г); зависит она от природы и свойств ионита.
Ввиду особой роли иона Н+(ОН-) и присутствия его в любом водном растворе общее количество поглощенных остальных катионов (анионов) зависит от рН, аi и ионного состава. Поэтому целесообразно ввести величину g, характеризующую поглощение ионов в определенных стандартных условиях —условную емкость обмена.
Условной емкостью обмена называют число моль-экв солевых ионов, поглощенных 1 кг ионита при заданных значениях рН, концентрации и состава раствора.
Значения g для почв и неорганических высокодисперсных ионитов обычно не превышают 1 моль-экв/кг. Синтетические ионообменные смолы характеризуются емкостью 3—10 моль-экв/кг и поэтому чаще всего используются в практике.
В настоящее время ионный обмен находит широкое практическое применение в самых различных отраслях промышленности.
Одно из наиболее важных применений ионного обмена — получение в производственных масштабах воды, пригодной для пищевых и технических целей. Все природные воды обладают большей или меньшей жесткостью, обусловленной присутствием ионов Са2+ и Mg2+. Жесткая вода не может быть использована в паровых котлах (из-за образования накипи). Она нарушает моющее действие мыл, неприменима для многих производственных процессов и часто непригодна для питья и приготовления пищи.
Разнообразные способы умягчения воды разрабатывались с давних времен, однако лишь с использованием ионного обмена удалось осуществить достаточно рентабельный метод умягчения воды в промышленных масштабах. В этом методе ионообменную смолу (катионит), выпускаемую промышленностью обычно в виде зерен, переводят в Nа+-форму и загружают в колонку, через которую пропускают природную воду. Процесс идет по схеме
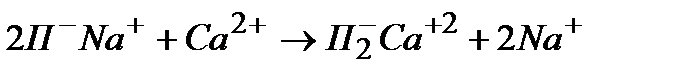
Весьма важной является также задача частичного или полного опреснения (обессоливания) воды, т. е. удаления содержащихся в ней электролитов (получение пресной воды на морских судах и т. д.). Для этого воду последовательно пропускают через Н+-форму катионита сильно кислотного типа и затем — через ОН--форму сильного анионита:
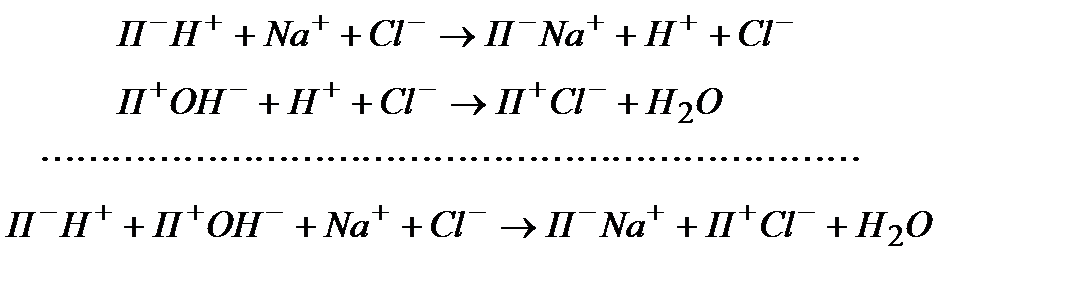
Иониты после использования их емкости могут быть легко регенерированы посредством обработки кислотой и щелочью. Поскольку последние легко получаются из природной воды электролизом, процесс опреснения, в принципе, не требует расхода химических веществ, — затрачивается лишь электроэнергия. Промышленные иониты обладают высокой механической и химической стойкостью и выдерживают практически сотни регенерационных циклов.
Подобные же схемы применяются для решения одной из важнейших проблем современности — очистки заводских сточных вод, в которых многие вредные вещества содержатся в ионной форме (ионы тяжелых металлов, органические основания и др.). Задача извлечения этих веществ, часто весьма ценных для промышленности, решается во многих случаях сравнительно легко, благодаря их высокой адсорбционной способности, применением рассмотренных схем.
Необходимо упомянуть о применении ионного обмена в современной медицине: при заболеваниях, характеризующихся нарушениями ионного баланса в органах и тканях (язве желудка, гипертонических отеках и др.). Введением высокодиспергированных порошков из ионообменных смол удается во многих случаях сдвинуть в нужную сторону и далее поддерживать необходимый ионный баланс организма.
* Электрофорез - явление перемещения частиц дисперсной фазы в электрическом поле.
Дата добавления: 2017-10-09; просмотров: 2924;