Нарушения метаболизма галактозы
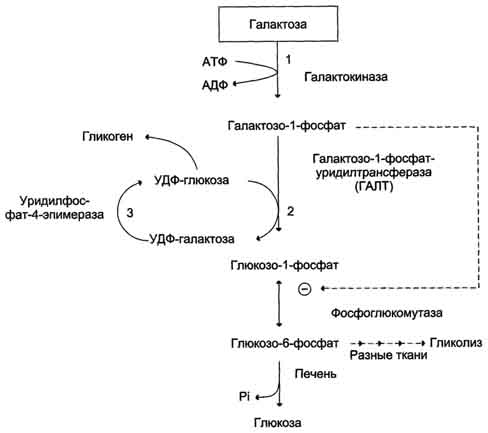
Обмен галактозы особенно интересен в связи с наследственным заболеванием - галактоземией. Галактоземиявозникает при нарушении обмена галактозы, обусловленном наследственным дефектом любого из трёх ферментов, включающих галактозу в метаболизм глюкозы
| Дефектный фермент (частота) | Блокируемая реакция | Клинические проявления и лабораторные данные |
| Галактокиназа (1:500 000) | Галактоза + АТФ → Галактозо-1-фосфат + АДФ | Галактоземия, галактозурия, катаракта. Активность фермента в эритроцитах нормальная. |
| Галактозо-1-фосфатуридилтрансфераза (1:40000) | Галактозо-1-фосфат + УДФ-глюкоза → УДФ-галактоза + Глюкозо-1-фосфат | Галактоземия, галактозурия, галактозо-1-фосфатемия, катаракта. Тенденция к гипогликемии, компенсаторная мобилизация жиров, цирроз печени, нарушения функции почек. Гепатомегалия, задержка психического развития. Активность фермента в эритроцитах снижена. |
| Уридилфосфат-4-эпимераза (1:1000000) | УДФ-глюкоза ↔ УДФ-галактоза | Галактоземия, галактозурия. Тяжёлых клинических проявлений нет. Описаны единичные случаи заболевания. |
Галактоземия, вызванная недостаточностью галактозо-1-фосфатуридилтрансферазы (ГАЛТ), наиболее хорошо изучена. Это заболевание проявляется очень рано, и особенно опасно для детей, так как основным источником углеводов для них служит материнское молоко, содержащее лактозу. Ранние симптомы дефекта ГАЛТ: рвота, диарея, дегидратация, уменьшение массы тела, желтуха. Они появляются вскоре после рождения, как только ребёнок начинает получать молоко. В крови, моче и тканях повышается концентрация галактозы и галактозо-1-фосфата. В тканях глаза (в хрусталике) галактоза восстанавливается альдоредуктазой с образованием галактитола (дульцита). В этой реакции в качестве донора водорода используется NADPH. Восстановление галактозы происходит и в ходе нормального метаболизма, но протекает с небольшой скоростью. При галактоземии галактитол накапливается в стекловидном теле и связывает большое количество воды. Вследствие этого нарушается баланс электролитов, а чрезмерная гидратация хрусталика приводит к развитию катаракты, которая наблюдается уже через несколько дней после рождения.
Тяжёлые последствия дефекта ГАЛТ наблюдают в печени. Это связано с накоплением галактозо-1-фосфата и его токсическим действием на гепатоциты. В результате возникают нарушения функции печени: гепатомегалия, жировая дистрофия. В почках таких больных также повышена концентрация галактитола и галактозо-1-фосфата, что влияет на их функции. Отмечают нарушения в клетках полушарий головного мозга и мозжечка, в тяжёлых случаях - отёк мозга, задержку умственного развития, возможен летальный исход. Для галактоземии, вызванной дефектом галактокиназы, тоже характерна катаракта, но при этом заболевании, в отличие от дефекта ГАЛТ, не отмечают нарушений функций печени, почек, мозга. Наиболее тяжёлые последствия снижения активности ГАЛТ связывают с влиянием галактозо-1-фосфата на активность других ферментов, участвующих в углеводном обмене (фосфоглюкомутазы, глюкозо-6-фосфатдегидрогеназы). Известно несколько форм галактоземии, причиной которой является недостаточность ГАЛТ Некоторые дефекты в строении ГАЛТ приводят лишь к частичной потере активности фермента. Поскольку в норме ГАЛТ присутствует в организме в избытке, то снижение его активности до 50%, а иногда и ниже может клинически не проявляться. При диагностике галактоземии исследуют мочу на содержание галактозы, собранную после нескольких кормлений молоком. При обнаружении у ребёнка катаракты его обследуют на недостаточность галактокиназы и ГАЛТ. Наличие галактозы в моче при отсутствии нарушений функции печени указывает на дефект галактокиназы. При обследовании проведение теста с нагрузкой галактозой не рекомендуется, так как этот тест опасен для больных. Лечение заключается в удалении галактозы из рациона.
Классификация, биохимическая и генетическая характеристика гликогенов.
| Тип и синонимы | Дефектный фермент | Наследование | Структура и особенности накопления гликогена | Клинические проявления |
| 0 (недостаточность гликосинтетазы) | Гликосинтетаза | Аутосомно -рецессивное | Нормальная структура, пониженное содержание | Тяжелая гипогликемия с кетоацидозом утром натощак и гипергликемия с лактацидозом днем после еды. |
| I (болезнь Гирке) | Ферментная система печени, превращающая глюкозо-6-фосфат в глюкозу; тип Iа: глюкозо-6- фосфатаза, тип Ib: глюкозо-6- фосфат-транслоказа | Аутосомно -рецессивное | Нормальная структура, повышенное содержание | гипогликемия голодания, гепатомегалия, дистрофия мышц, задержка роста, лактоацидоз, ожирение. |
| II (болезнь Помпе) | Лизосомальная a-D-глюкозидаза | Аутосомно -рецессивное | Нормальная структура, повышенное содержание, гликоген накапливается в лизосомах | Накопление гликогена в мышцах |
| III (болезнь Форбса, болезнь Кори, болезнь остаточного декстрина) | Амило-1,6- глюкозидаза, 4- a-D- глюканотрансфераза | Аутосомно -рецессивное | Короткие боковые цепи, повышенное содержание | выраженная кетонемия, миопатия, спленомегалия |
| IV (болезнь Андерсен, амилопектиноз0 | 1,4-a-глюкан-ветвящий фермент | Аутосомно -рецессивное | Удлиненные боковые цепи, нормальное содержание | - |
| V (болезнь МакАрдля, недостаточность мышечной фосфорилазы) | Фосфорилаза в мышцах | Аутосомно -рецессивное | Нормальная структура, умеренно повышенное содержание | - |
| VI (болезнь Герса) | Фосфорилаза в печени | Аутосомно -рецессивное | Нормальная структура, повышенное содержание | - |
| VII (болезнь Таруи) | Мышечная фосфофруктокиназа | Аутосомно -рецессивное | Нормальная структура, повышенное содержание | - |
58. Важнейшие липиды тканей человека. Резервные липиды (жиры) и липиды мембран (сложные липиды). Жирные кислоты липидов тканей человека.
Жирные кислоты- структурные компоненты различных липидов. В составе триацилгли-церолов жирные кислоты выполняют функцию депонирования энергии, так как их радикалы содержат богатые энергией СН2-группы. При окислении СН-связей энергии выделяется больше, чем при окислении углеводов, в которых атомы углерода уже частично окислены (-НСОН-). В составе фосфолипидов и сфинго-липидов жирные кислоты образуют внутренний гидрофобный слой мембран, определяя его свойства. Жиры и фосфолипиды организма при нормальной температуре тела имеют жидкую консистенцию, так как количество ненасыщенных жирных кислот преобладает над насыщенными. В фосфолипидах мембран ненасыщенных кислот может быть до 80-85%, а в составе жиров подкожного жира - до 60%. Жирные кислоты липидов человека представляют собой углеводородную неразветвлённую цепь, на одном конце которой находится карбоксильная группа, а на другом - метальная группа (ω-углеродный атом). Большинство жирных кислот в организме содержат чётное число атомов углерода - от 16 до 20
Дата добавления: 2016-12-26; просмотров: 992;