Механизм токсического действия и патогенез интоксикации метилового спирта, этиленгликоля и дихлорэтана.
Метиловый спирт.
После приема внутрь метанол быстро всасывается и распределяется в биосредах. Средние значения смертельных концентраций яда в плазме у взрослых составляют 1 г/л, у детей – 0,4 г/л. Метанол преимущественно разрушается в печени (94%), 5% выводится почками в неизмененном виде, 1% – с выдыхаемым воздухом. Период полувыведения метанола (T05), принятого в низких дозах, составляет 14–27 ч и увеличивается до 30 ч при приеме в высоких дозах.
Метаболизм метанола изучен достаточно подробно. Основными метаболитами метанола являются формальдегид и муравьиная кислота, причем трансформация формальдегида в формиат происходит быстро, а расщепление муравьиной кислоты до углекислого газа и воды весьма медленно. Это приводит к тому, что в биосредах накапливаются значительные количества формиата. Биологическое действие неизмененной молекулы метанола ограничивается наркотическим эффектом. Токсичность метилового спирта обусловлена формальдегидом и муравьиной кислотой. Эти метаболиты оказывают многостороннее действие на биохимические системы организма.
Главные направления их действия:
1. подавление окислительного фосфорилирования с развитием дефицита АТФ;
2. метаболический ацидоз (как за счет нарушения окисления, так и в результате накопления формиата);
3. снижение уровня восстановленного глутатиона, дефицит сульфгидрильных групп;
4. образование конъюгатов с биологически активными веществами – аминами, вазоактивными соединениями, нейромедиаторами, нуклеотидами и др.
Метанол – сильный нейрососудистый яд. Основными объектами его воздействия являются наиболее чувствительные к недостатку АТФ структуры (головной мозг, сетчатка и зрительный нерв). Окулотоксическое действие проявляется в различные сроки после приема яда (от 40 мин до 72 ч). При офтальмоскопии регистрируют отек диска зрительного нерва, который развивается вследствие его демиелинизации.
В основе повреждений органа зрения лежат нарушения фосфорилирующих процессов в системе цитохромоксидазы (цитохром AA3). В результате нарушается энергообразование, и как следствие – изменение массопереноса веществ через аксолемму, что приводит к демиелинизации и последующей атрофии зрительного нерва в целом.
Поражения усугубляются метаболическим ацидозом, нарушениями обмена вазоактивных веществ и нейромедиаторов, расстройствами общей и церебральной гемодинамики, повышением проницаемости мембран, перераспределением жидкости с развитием отека головного мозга.
Общемозговые расстройства с нарушением жизненно важных функций являются основной причиной смерти отравленных метанолом.
Этиленгликоль
Этиленгликоль быстро всасывается в желудочно‑кишечном тракте, определяется в крови через 5–15 мин после приема внутрь и достигает максимальной концентрации в крови через 2–5 ч. В связи с этим необходимо у таких больных как можно раньше промыть желудок и очистить кишечник, что и объясняет наибольшую эффективность применения в эти сроки методов форсированного удаления невсосавшегося яда из организма. Этиленгликоль и продукты его распада можно обнаружить в организме до 3–8 дней и более.
Трансформация этиленгликоля в организме происходит по следующей схеме:
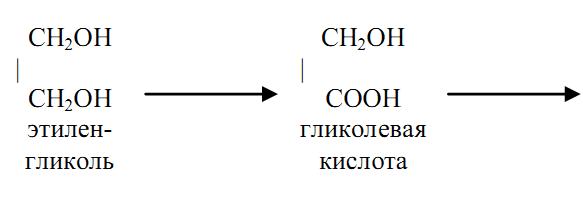
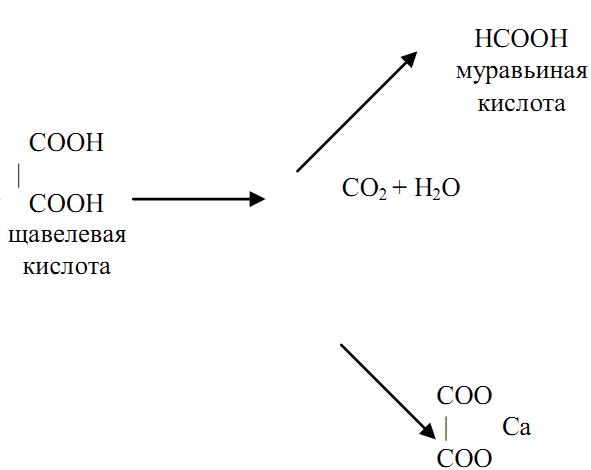
Этиленгликоль подвергается в организме окислению до углекислоты и воды с образованием токсичных промежуточных продуктов – гликолевого альдегида, гликолевой кислоты, щавелевой, уксусной, муравьиной и других кислот, что приводит к ацидозу, выраженным расстройствам обмена, гипоксии. Щавелевая кислота взаимодействует с ионами кальция с образованием нерастворимой щавелевокислой кальциевой соли. Основной путь удаления из организма этиленгликоля и продуктов его распада‑ через почки (до 50% яда). При этом оксалаты осаждаются в стенках капилляров, в канальцах и лоханках почек и, действуя непосредственно и рефлекторным путем, нарушая почечный кровоток, способствуют развитию токсической нефропатии. Гипокальциемия, вызванная связыванием ионизированного кальция, может быть одной из причин нарушения деятельности центральной нервной системы и работы сердца.
Этиленгликоль действует как сосудистый и протоплазматический яд, подавляет окислительные процессы, вызывает отек, набухание и некроз мелких сосудов с расстройством тканевого кровообращения, сдвигает кислотно‑щелочное равновесие в сторону метаболического ацидоза, нарушает водно‑электролитный баланс.
В механизме токсического действия этиленгликоля определенная роль отводится как неизмененному гликолю, так и продуктам его биотрансформации. С целой молекулой связано умеренно выраженное наркотическое действие яда, а также высокая осмотичность, вследствие чего возможны водная дегенерация клетки почечного эпителия и отек мозга. Ведущая же роль в развитии отравления принадлежит метаболитам этиленгликоля. В течение длительного времени основное значение придавалось щавелевой кислоте, способной связывать кальций с образованием плохо растворимого оксалата.
Однако оказалось, что в оксалат трансформируется лишь незначительная доля этиленгликоля, а гипокальциемия развивается далеко не во всех случаях тяжелых отравлений. С другой стороны, кристаллы кальция оксалата образуются в почках, мозге и легких, что ухудшает функцию этих органов.
В настоящее время считается, что в формировании цитотоксического эффекта этиленгликоля главную роль играет гликолевая кислота и ее метаболит – глиоксиловая кислота, которая наиболее токсична. Она разобщает окисление и фосфорилирование.
Таким образом, продукты биотрансформации этиленгликоля вызывают серьезные и разнообразные нарушения энзиматических процессов. Указанные нарушения усиливаются вследствие осмотического действия яда, а также метаболического ацидоза, развивающегося в результате накопления эндогенных продуктов и кислот, образующихся при метаболизме этиленгликоля.
Метаболические расстройства являются пусковым звеном в развитии поражений, наиболее выраженных в головном мозге, почках и печени. Тяжелые расстройства обмена веществ, гипоксия, повышение мембранной проницаемости способствуют формированию экзотоксического шока.
Особенно значительные гемодинамические расстройства при отравлениях этиленгликолем наблюдаются в почках. Известно, что при снижении ОЦК в 2 раза почечный кровоток уменьшается в 20 – 30 раз.
Замедление почечного кровотока, стазы, повышение проницаемости мембран приводят к ишемии ткани почек, отеку интерстиция, повышению внутриорганного давления, нарушению фильтрационно‑реабсорбционных процессов, прогрессированию расстройств гемо‑ и лимфотока.
Рефлекторный спазм артерий коры, раскрытие артерио‑ венозных анастомозов со сбросом крови через юкстамедуллярные пути еще более усиливают поражения почечной паренхимы.
Указанные нарушения в сочетании с прямым повреждающим действием продуктов метаболизма этиленгликоля приводят к развитию весьма характерного для данной интоксикации тотального двухстороннего коркового некроза почек.
Дихлорэтан.
Токсическое действие дихлорэтана связано с продуктами его биотрансформации. Так, в процессе дехлорирования образуется 1‑ хлорэтанол, который при участии алкоголь‑ и альдегиддегидрогеназы окисляется до хлорацетоальдегида и монохлоруксусной кислоты. Естественный путь детоксикации дихлорэтана в организме, как и других рассматриваемых углеводородов, – это реакция с восстановленным глутатионом в печени; в результате образуются малотоксичные меркаптуровые кислоты, однако один из промежуточных продуктов – полуиприт – способен оказывать алкилирующее действие. Метаболиты дихлорэтана обладают высокой активностью и, вступая во взаимодействие с сульфгидрильными группами ферментов, нарушают их структуру и функцию. Наибольшей токсичностью обладает, по‑видимому, монохлоруксусная кислота, которая, блокируя акониттрансферазу, дезорганизует работу цикла трикарбоновых кислот.
Токсичные метаболиты хлорорганических соединений путем алкилирования и (или) стимуляции перекисного окисления липидов повреждают плазматические и внутриклеточные мембраны и запускают, судя по всему, кальциевый механизм гибели клеток. Внутриклеточное накопление кальция блокирует митохондриальное окислительное фосфорилирование, дестабилизирует лизосомальные мембраны, активирует находящиеся в лизосомах эндопротеазы, обладающие аутопротеолитическими свойствами. Следствием этих изменений, а также расстройств липидного обмена (увеличение количества липидов, поступающих в клетку, и угнетение их выведения), являются дистрофические (преимущественно жировая дистрофия) и некротические поражения клеток.
Указанные механизмы (неэлектролитное и электролитное действие токсиканта) являются первичными, реализующимися уже в токсикогенной стадии интоксикации. Они вызывают изменения в различных органах и тканях, приводят к серьезным расстройствам гомеостаза (метаболическому ацидозу, водно‑электролитным, гемокоагуляционным сдвигам и т. д.), формированию ряда вторичных синдромов (центральных и аспирационно‑обтурационных нарушений дыхания, острой недостаточности паренхиматозных органов и т. д.).
Важное место в патогенезе интоксикации хлорорганическими соединениями занимают расстройства гемодинамики, особенно экзотоксический шок, – следствие резкого увеличения проницаемости сосудистой стенки с выходом жидкой части крови в интерстиций, развитием истинной гиповолемии, централизацией кровообращения, периферической вазоконстрикцией, гемоконцентрацией, агрегацией форменных элементов, значительными нарушениями микроциркуляции, углубляющими гипоксию тканей и нарушения гомеостаза.
Описанные выше нарушения на определенном этапе формирования экзотоксического шока приводят к выраженным изменениям реологических свойств крови с развитием в дальнейшем коагулопатии потребления (ДВС‑синдрома).
В соматогенной стадии интоксикации главное место занимают поражения паренхиматозных органов – печени и почек. Дистрофические и некротические изменения в клетках этих органов сопровождаются нарушением всех основных функций печени – синтетической, детоксикационной, регулирующей все основные виды межуточного обмена, и почек – выделения воды, электролитов, азотистых шлаков, регуляции гемопоэза, артериального давления. Эти метаболические нарушения, а также продукты деструкции собственно паренхиматозных органов являются основой формирования вторичной эндогенной интоксикации, нередко с проявлениями полиорганной недостаточности, которая сама по себе приводит к нарастанию дегенеративно‑дистрофических изменений в тканях, способствует развитию осложнений, в том числе инфекционных.
Дата добавления: 2016-02-02; просмотров: 4161;