Глава 22. Метаболизм холестерола. Биохимия атеросклероза
Холестерол – стероид, характерный только для животных организмов. Основное место его образования в организме человека – печень, где синтезируется 50% холестерола, в тонком кишечнике его образуется 15–20%, остальное количество синтезируется в коже, коре надпочечников и половых железах. Источники формирования фонда холестерола и пути его расходования представлены на рис 22.1.
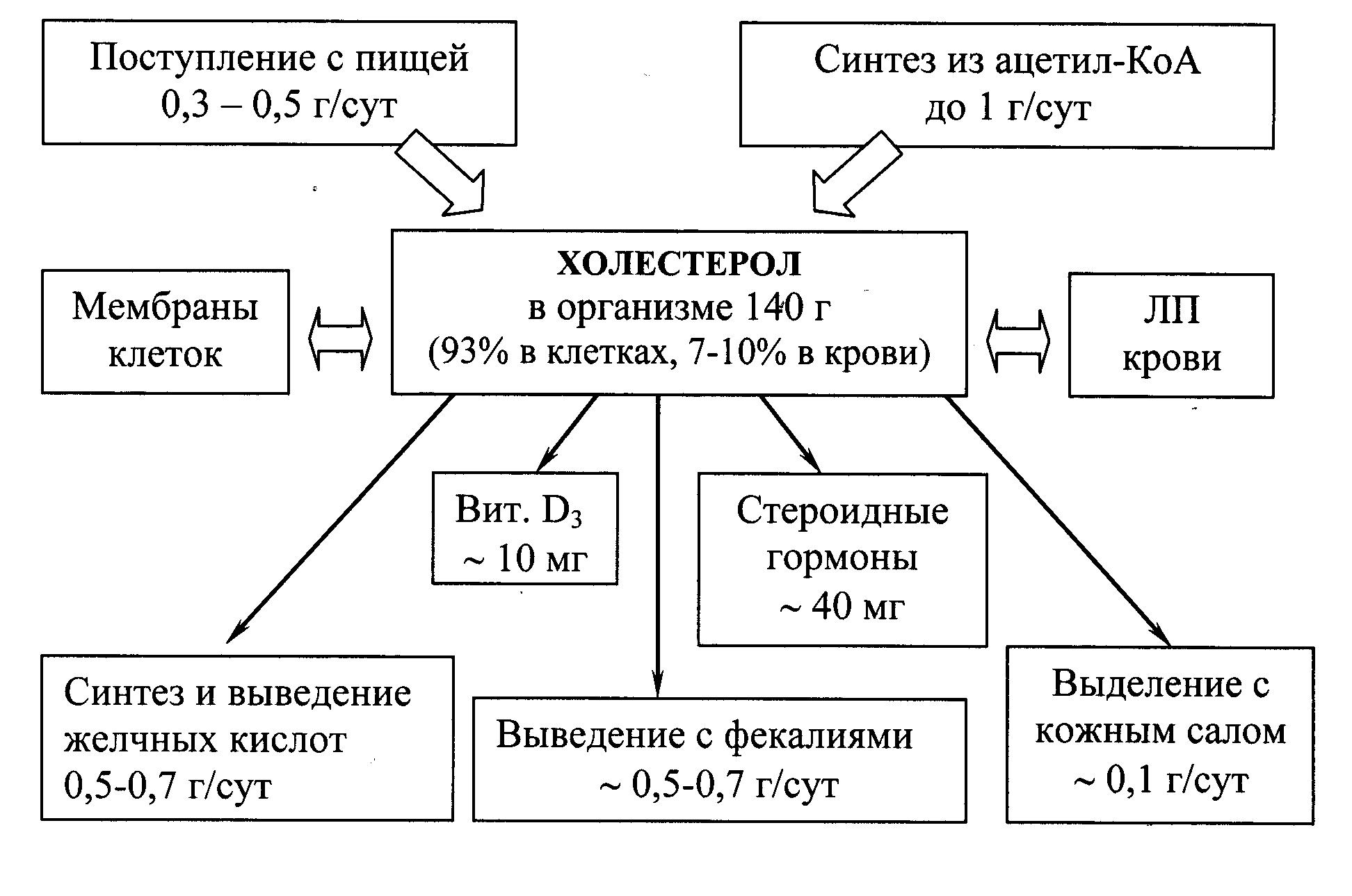
Рис. 22.1. Формирование и распределение фонда холестерола в организме.
Холестерол организма человека (суммарное количество около 140 г) условно можно разделить на три пула:
1. пул А (~ 30 г), быстрообменивающийся, состоит из ХС кишечной стенки, плазмы крови, печени и других паренхиматозных органов, обновление происходит за 30 сут (1 г/сут);
2. пул Б (~ 50 г), медленнообменивающийся ХС остальных органов и тканей;
3. пул В (~ 60 г), очень медленнообменивающийся ХС спинного и головного мозга, соединительной ткани, скорость обновления исчисляется годами.
Синтез холестерола происходит в цитозоле клеток. Это один из самых длинных метаболических путей в организме человека. Он проходит в 3 этапа: первый заканчивается образованием мевалоновой кислоты, второй – образованием сквалена (углеводород линейной структуры, состоящий из 30 углеродных атомов). В ходе третьего этапа сквален превращается в молекулу ланостерола, далее происходит 20 последовательных реакций, превращающих ланостерол в холестерол.
В некоторых тканях гидроксильная группа холестерола этерифицируется с образованием эфиров. Реакция катализируется внутриклеточным ферментом АХАТ (ацилКоА:холестеролацилтрансферазой). Реакция этерификации происходит также в крови в ЛПВП, где находится фермент ЛХАТ (лецитин:холестеролацилтрансфераза). Эфиры холестерола – форма, в которой он транспортируется кровью или депонируется в клетках. В крови около 75% ХС находится в виде эфиров.
Регуляция синтеза холестерола осуществляется путем влияния на активность и количество ключевого фермента процесса – 3‑гидрокси‑3‑метилглутарил‑КоА‑редуктазы (ГМГ‑КоА‑редуктазы).
Это достигается двумя способами:
1. Фосфорилирование/дефосфорилирование ГМГ‑КоА‑редуктазы. Инсулин стимулирует дефосфорилирование ГМГ‑КоА‑редуктазы, переводя её тем самым в активное состояние. Следовательно, в абсорбтивный период синтез ХС увеличивается. В этот период увеличивается и доступность исходного субстрата для синтеза – ацетил‑КоА. Глюкагон оказывает противоположное действие: через протеинкиназу А стимулирует фосфорилирование ГМГ‑КоА‑редуктазы, переводя её в неактивное состояние. В результате синтез ХС в постабсорбтивном периоде и при голодании ингибируется.
2. Ингибирование синтеза ГМГ‑КоА‑редуктазы. ХС (конечный продукт метаболического пути) снижает скорость транскрипции гена ГМГ‑КоА‑редуктазы, подавляя таким образом собственный синтез, аналогичный эффект вызывают и жёлчные кислоты.
Транспорт холестерола кровью осуществляется в составе ЛП. ЛП обеспечивают поступление в ткани экзогенного ХС, определяют его потоки между органами и выведение из организма. Экзогенный ХС доставляется в печень в составе остаточных ХМ. Там вместе с синтезированным эндогенным ХС он формирует общий фонд. В гепатоцитах ТАГ и ХС упаковываются в ЛПОНП, и в таком виде секретируются в кровь. В крови ЛПОНП под действием ЛП‑липазы, гидролизующей ТАГ до глицерола и жирных кислот, превращаютя сначала в ЛППП, а затем и в ЛПНП, содержащие до 55% ХС и его эфиров. ЛПНП – основная транспортная форма ХС, в которой он доставляется в ткани (70% ХС и его эфиров в крови находится в составе ЛПНП). Из крови ЛПНП поступают в печень (до 75%) и другие ткани, которые имеют на своей поверхности рецепторы ЛПНП.
Если количество ХС, поступающего в клетку, превышает её потребность, то синтез рецепторов ЛПНП подавляется, что уменьшает поток ХС из крови. При снижении концентрации свободного ХС в клетке, наоборот, синтез рецепторов активируется. В регуляции синтеза рецепторов ЛПНП участвуют гормоны: инсулин, трийодтиронин и половые гормоны увеличивают образование рецепторов, а глюкокортикоиды – уменьшают.
В так называемом «обратном транспорте холестерола», т.е. пути, обеспечивающем возвращение ХС в печень, основную роль играют ЛПВП. Они синтезируются в печени в виде незрелых предшественников, которые практически не содержат ХС и ТАГ. В крови предшественники ЛПВП насыщаются ХС, получая его из других ЛП и мембран клеток. В переносе ХС в ЛПВП участвует фермент ЛХАТ, находящийся на их поверхности. Этот фермент присоединяет остаток жирной кислоты от фосфатидилхолина (лецитина) к ХС. В результате образуется гидрофобная молекула эфира холестерола, которая перемещается внутрь ЛПВП. Таким образом, незрезые ЛПВП, обогащаясь ХС, превращаются в ЛПВП3 – зрелые и более крупные по размерам частицы. ЛПВП3 обменивают эфиры холестерола на ТАГ, содержащиеся в ЛПОНП и ЛППП при участии специфического белка, переносящего эфиры холестерола между липопротеинами. При этом ЛПВП3 превращаются в ЛПВП2, размер которых увеличивается за счет накопления ТАГ. ЛПОНП и ЛППП под действием ЛП‑липазы превращаются в ЛПНП, которые в основном и доставляют ХС в печень. Небольшая часть ХС доставляется в печень ЛПВП2 и ЛППП.
Синтез жёлчных кислот. В печени из ХС синтезируется 500–700 мг жёлчных кислот в сутки. Их образование включает реакции введения гидроксильных групп при участии гидроксилаз и реакции частичного окисления боковой цепи ХС (Рис. 22.2):
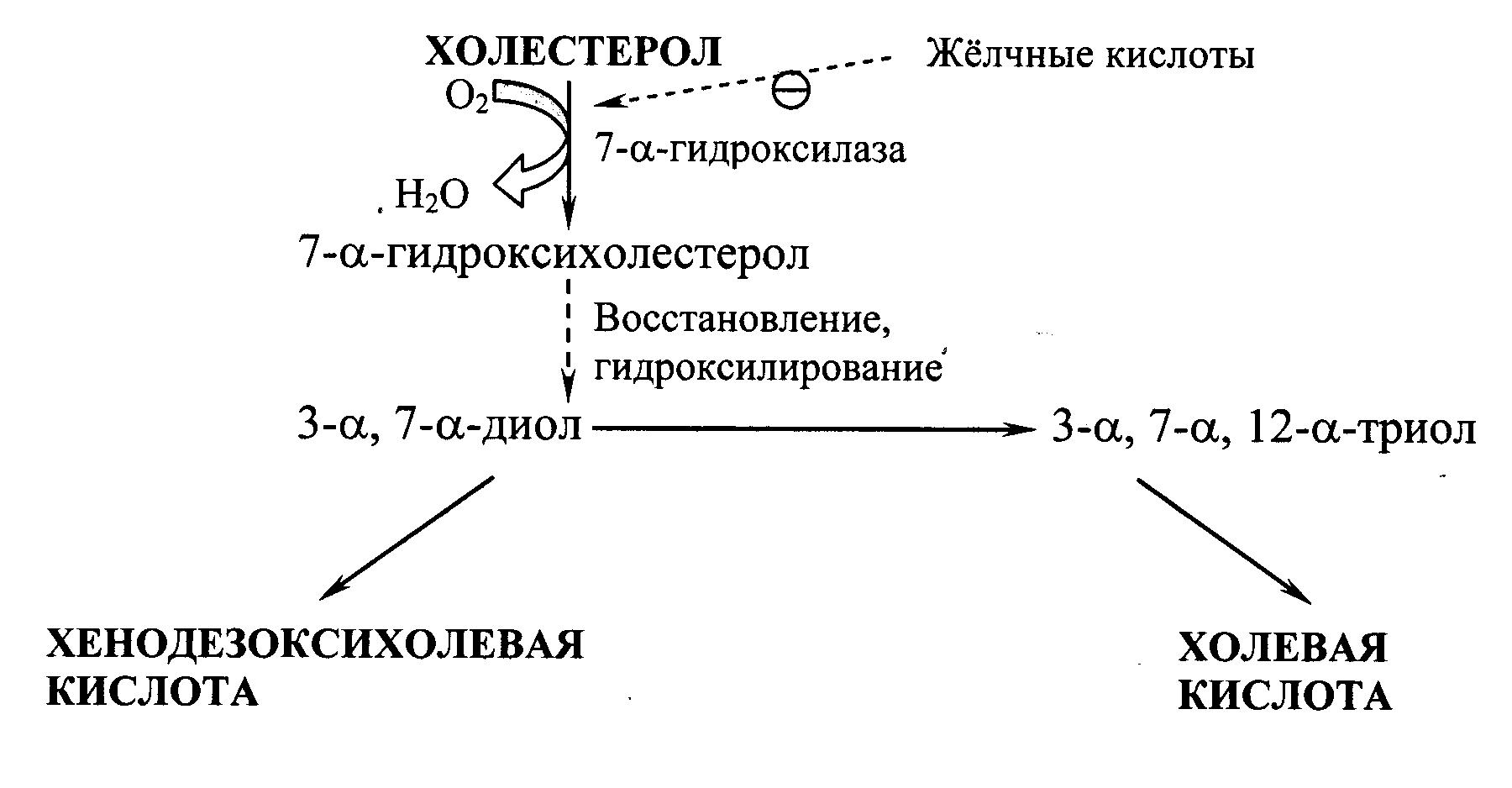
Рис. 22.2. Схема образования жёлчных кислот.
Первая реакция синтеза – образование 7‑a‑гидроксихолестерола – является регуляторной. Активность фермента, катализирующего эту реакцию, ингибируется конечным продуктом пути – жёлчными кислотами. Еще одним механизмом регуляции является фосфорилирование/дефосфорилирование фермента (активна фосфорилированная форма 7‑a‑гидроксилазы). Возможна и регуляция путем изменения количества фермента: ХС индуцирует транскрипцию гена 7‑a‑гидроксилазы, а жёлчные кислоты репрессируют. Тиреоидные гормоны индуцируют синтез 7‑a‑гидроксилазы, а эстрогены – репрессируют. Такое влияние эстрогенов на синтез жёлчных кислот объясняет, почему желчнокаменная болезнь встречается у женщин в 3–4 раза чаще, чем у мужчин.
Образовавшиеся из ХС холевую и хенодезоксихолевую кислоты называют «первичными жёлчными кислотами». Основная масса этих кислот подвергается коньюгации – присоединению молекул глицина или таурина к карбоксильной группе жёлчной кислоты. Коньюгация начинается с образования активной формы желчных кислот – производных КоА, затем присоединяются таурин или глицин, и в результате образуется 4 варианта коньюгатов: таурохолевая и таурохенодезоксихолевая, гликохолевая и гликохенодезоксихолевая кислоты. Они являются значительно более сильными эмульгаторами, чем исходные жёлчные кислоты. Коньюгатов с глицином образуется в 3 раза больше, чем с таурином, так как количество таурина в организме ограничено. В кишечнике небольшое количество коньюгатов первичных жёлчных кислот под действием ферментов бактерий превращаются во вторичные жёлчные кислоты. Дезоксихолевая кислота, образующаяся из холевой, и литохолевая, образующаяся из дезоксихолевой, хуже растворимы и медленнее всасываются в кишечнике.
Около 95% жёлчных кислот, попавших в кишечник, возвращаются в печень через воротную вену, затем опять секретируются в жёлчь и повторно используются в эмульгировании жиров. Этот путь жёлчных кислот называется энтерогепатической циркуляцией. С фекалиями в основном удаляются вторичные жёлчные кислоты.
Желчнокаменная болезнь (ЖКБ) – патологический процесс, при котором в жёлчном пузыре образуются камни, основу которых составляет ХС.
Выделение ХС в жёлчь должно сопровождаться пропорциональным выделением жёлчных кислот и фосфолипидов, удерживающих гидрофобные молекулы ХС в мицеллярном состоянии. Причинами, приводящими к изменению соотношения жёлчных кислот и ХС в жёлчи являются: пища, богатая ХС, высококалорийное питание, застой жёлчи в жёлчном пузыре, нарушение энтерогепатической циркуляции, нарушения синтеза жёлчных кислот, инфекции жёлчного пузыря.
У большинства больных ЖКБ синтез ХС увеличен, а синтез жёлчных кислот из него замедлен, что приводит к диспропорции количества ХС и жёлчных кислот, секретируемых в жёлчь. В итоге ХС начинает осаждаться в жёлчном пузыре, образуя вязкий осадок, который постепенно затвердевает. Иногда он пропитывается билирубином, белками и солями кальция. Камни могут состоять только из ХС (холестериновые камни) или из смеси ХС, билирубина, белков и кальция. Холестериновые камни обычно белого цвета, а смешанные – коричневые разных оттенков.
В начальной стадии образования камней можно применять в качестве лекарства хенодезоксихолевую кислоту. Попадая в жёлчный пузырь, она постепенно растворяет холестериновые камни, однако это медленный процесс, длящийся несколько месяцев.
Дата добавления: 2016-01-30; просмотров: 1371;