Нарушения сердечного ритма и проводимости (с указанием формы). 22 страница
К эссенциальной (первичной) АГ относят те случаи заболевания, когда невозможно установить связь между повышением АД и той или иной органной или эндокринной патологией, предшествующей возникновению АГ.
На долю эссенциальной АГ приходится около 90–95% случаев хронического повышения уровня АД. В России для обозначения первичной АГ неизвестной этиологии традиционно употребляют другой термин — “гипертоническая болезнь”, который является синонимом термина “эссенциальная АГ”. Диагноз гипертонической болезни (ГБ) ставят фактически после исключения того или иного варианта симптоматических АГ.
В данной главе будут рассмотрены вопросы этиологии, патогенеза, клиники, диагностики и лечения наиболее распространенной формы АГ — гипертонической болезни.
| 7.1. Этиология |

|
Хотя этиология эссенциальной АГ (ГБ) остается неизвестной, в настоящее время уже хорошо изучены некоторые неблагоприятные факторы (факторы риска), предрасполагающие к развитию ГБ, а также многие патогенетические механизмы формирования заболевания. К числу наиболее значимых из них относятся следующие.
1. Наследственная предрасположенность играет важную роль в происхождении ГБ. Доказано, что лица, родители которых страдали ГБ, имеют более высокий риск развития этого заболевания и более высокую смертность от сердечно-сосудистых болезней. Причем имеет значение не столько сам факт наличия АГ у ближайших родственников, сколько характер и тяжесть течения заболевания, возраст, в котором впервые было отмечено повышение АД, а также наличие у родственников тяжелых сосудистых осложнений ГБ. Все же следует иметь в виду, что имеющаяся генетическая предрасположенность к возникновению АГ, как правило, реализуется только под действием некоторых неблагоприятных факторов окружающей среды и изменения образа жизни современного человека. По-видимому, генетически зашифрованными оказываются лишь органные, биохимические, клеточные и другие особенности ответа системы кровообращения на воздействия разнообразных внешних факторов.
2. Ограничение физической активности (гиподинамия). Эта важнейшая особенность современного образа жизни большинства жителей экономически развитых стран приводит, как известно, к детренированности организма и резкому снижению адаптационных возможностей не только мышечной системы, но и систем кровообращения, дыхания и др. В этих условиях повседневные жизненные ситуации (умеренная физическая нагрузка, психоэмоциональное напряжение и др.) вызывают чрезмерный (гиперреактивный) ответ в виде тахикардии, повышения АД и других неадекватных реакций, происходящих на системном, органном и клеточном уровнях. Такая дезадаптация и является, вероятно, первоначальной причиной постепенного формирования ГБ.
3. Ожирение способствует, как известно, существенному (в 2–6 раз) увеличению риска развития АГ (ВОЗ, 1996). Существует линейная зависимость уровня АД от массы тела, что объясняется, прежде всего, частотой выявления при ожирении так называемого метаболического синдрома, лежащего в основе выраженных нарушений эндотелиальной функции, когда в местной регуляции сосудистого тонуса начинают преобладать прессорные стимулы, что и приводит к прогрессирующему повышению АД. Кроме того, имеют значение характерные для больных ожирением гиперлипидемии, ассоциирующиеся с частым атеросклеротическим поражением артерий, что также способствует повышению ригидности сосудистой стенки и извращенным вазоконстрикторным реакциям на физиологические внешние раздражители.
4. Избыточное потребление поваренной соли (NаСl) в некоторых случаях имеет решающее значение в генезе ГБ (например, “солевой” формы АГ). Доказано, что для взрослого человека адекватное поступление NаCl составляет 3,5–4,0 г соли в сутки (или около 60–70 мэкв натрия). Однако в экономически развитых странах, для которых характерна высокая заболеваемость АГ, реальное потребление соли в настоящее время составляет 6–18 г в сутки. В противоположность этому, у представителей некоторых народностей и племен, до сих пор ведущих изолированный от внешнего мира образ жизни (аборигены Новой Гвинеи и Полинезийских островов, эскимосы Аляски, индейцы Венесуэлы, сельское население Кении и др.), потребление соли не превышает 4 г в сутки, а повышение АД встречается крайне редко.
Избыточное поступление натрия (а также лития) с пищей приводит к увеличению осмотического давления, объема внеклеточной жидкости и ОЦК, что сопровождается возрастанием сердечного выброса и тенденцией к подъему АД. Кроме того, повышение концентрации внутриклеточного Nа+, согласно Nа+-Са2+–обменному механизму (см. главу 2), сопровождается увеличением внутриклеточной концентрации ионов Сa2+ и, соответственно, увеличением тонуса гладкой мускулатуры сосудистой стенки, что также ведет к подъему АД. Эти эффекты усугубляются уменьшением содержания ионов К+ в организме и ростом соотношения Nа+/К+.
5. Дефицит кальция и магния также способствует развитию АГ. Было показано, например, что у жителей, которые пользуются мягкой водой с низким содержанием солей, заболеваемость АГ и ИБС оказывается более высокой. Наоборот, при употреблении высокоминерализованной (жесткой) воды частота возникновения АГ уменьшается.
6. Чрезмерное потребление алкоголя приводит прежде всего к уменьшению чувствительности барорецепторов аорты и синокаротидной зоны, в связи с чем нарушается центральная регуляция АД.
7. Гиперлипидемия способствует структурно-функциональным изменениям артерий большого круга кровообращения (атеросклероз) и стабилизации повышенных цифр АД.
8. Курение также оказывает определенное влияние на уровень АД, прежде всего благодаря повреждению функции эндотелия и активации вазоконстрикторных эндотелиальных факторов (тканевой АII, эндотелин и др.).
9. Возраст относится к числу важнейших немодифицируемых факторов риска АГ. С возрастом снижается функциональная активность большинства регуляторных систем, обеспечивающих оптимальный уровень АД. Следует, однако, заметить, что эта тенденция в полной мере проявляется только у лиц, испытывающих все перечисленные выше воздействия окружающей среды и современного образа жизни, характерного для цивилизованных, экономически развитых стран Европы, Северной Америки и некоторых стран Азии. У упоминавшихся выше народностей и племен, ведущих примитивный образ жизни, прямой зависимости между уровнем АД и возрастом обнаружить не удается.
Таким образом, большинство из перечисленных неблагоприятных факторов риска, предрасполагающих к развитию АГ, связаны с коренным изменением образа жизни современного урбанизированного общества, в котором биологически запрограммированные системы адаптации приходят в противоречие с реальным их использованием организмом. Недаром эссенциальную АГ (ГБ) относят к “болезням ХХ века” или “болезням цивилизации”.
| 7.2.1. Основные механизмы повышения АД |

|
Уровень АД, как известно, определяется тремя основными гемодинамическими показателями:
1. Величиной сердечного выброса (МО), который в свою очередь зависит от сократимости миокарда ЛЖ, ЧСС, величины преднагрузки и других факторов.
2. Величиной общего периферического сопротивления (ОПСС), зависящей от тонуса сосудов мышечного типа (артериол), выраженности структурных изменений их сосудистой стенки, жесткости артерий эластического типа (крупных и средних артерий, аорты), вязкости крови и других параметров.
3. Объемом циркулирующей крови (ОЦК).
Соотношение этих трех гемодинамических показателей определяет уровень системного АД. В норме при повышении сердечного выброса снижается ОПСС, в частности, за счет уменьшения тонуса артерий мышечного типа. Наоборот, падение сердечного выброса сопровождается некоторым возрастанием ОПСС, что препятствует критическому снижению АД. Тот же эффект может быть достигнут за счет уменьшения натрийуреза и диуреза (задержка Nа+ и воды в организме) и увеличения ОЦК.
Изменение ОПСС в ту или другую сторону сопровождается соответствующим (но противоположным) изменением сердечного выброса и ОЦК. Например, при повышении АД за счет возрастания ОПСС увеличивается натрийурез и диурез и уменьшается ОЦК, что в физиологических условиях влечет за собой восстановление оптимального уровня АД.
Напомним, что контроль за соотношением трех гемодинамических показателей и уровнем АД обеспечивается сложной многоступенчатой системой регуляции, которая представлена следующими ее компонентами:
- центральным звеном регуляции (вазомоторным центром);
- артериальными баро- и хеморецепторами;
- симпатической и парасимпатической нервными системами, включая клеточные α- и β-адренорецепторы, М-холинорецепторы и т.д.;
- ренин-ангиотензин-альдостероновой системой (РААС);
- предсердным натрийуретическим фактором (ПНУФ);
- калликреин-кининовой системой;
- эндотелиальной системой местной регуляции сосудистого тонуса, включая NО, ЭГПФ, РGI2, эндотелин, АII и др.
Ясно, что любое нарушение этих и некоторых других механизмов регуляции, если оно сохраняется сравнительно длительное время, может привести к стойкому изменению соотношения МО, ОПСС и ОЦК и повышению АД.
Учитывая эти данные, можно предположить, что независимо от основного этиологического фактора формирование артериальной гипертензии возможно при нарушении соотношения трех описанных гемодинамических показателей (МО, ОПСС и ОЦК). Теоретически можно предположить следующие патогенетические варианты формирования эссенциальной АГ (ГБ):
1. АГ, обусловленная стойким повышением сердечного выброса, не сопровождающимся адекватным уменьшением ОПСС и ОЦК (например, за счет уменьшения сосудистого тонуса и натрийуреза).
2. АГ, вызванная преимущественным увеличением ОПСС без соответствующего снижения МО и ОЦК.
3. АГ, формирующаяся на фоне одновременного увеличения МО и ОПСС без адекватного снижения ОЦК (отсутствие адекватного увеличения натрийуреза).
4. АГ, обусловленная преимущественным увеличением ОЦК, связанным с резким уменьшением натрийуреза и диуреза (задержка натрия и воды в организме).
В реальной клинической практике перечисленные патогенетические варианты чаще всего являются лишь стадиями развития АГ у одного и того же больного, хотя в некоторых случаях преобладание одного из них может наблюдаться в течение всего заболевания.
Многообразие факторов, влияющих на уровень АД, объясняет всю сложность патогенеза ГБ и ее необычную полиэтиологичность. Существует мнение, что мы имеем дело не с одной, а с несколькими отдельными нозологическими единицами, объединяемыми в настоящее время термином “гипертоническая болезнь” на основании ведущего патогенетического признака — стойкого повышения системного АД (В.А. Люсов, В.И. Маколкин, Е.Н. Амосова и др.).
Это объясняет также существование множества гипотез этиологии и патогенеза эссенциальной АГ, каждая из которых не противоречит, а лишь дополняет наши представления о механизмах формирования и прогрессирования данного заболевания. На рис. 7.2, заимствованном из работы Dickinson (1991), представлены наиболее значимые механизмы регуляции АД, изучавшиеся на протяжении ХХ столетия, нарушение функционирования которых рассматривалось в качестве основной причины развития АГ. Коротко рассмотрим лишь некоторые из этих гипотез.
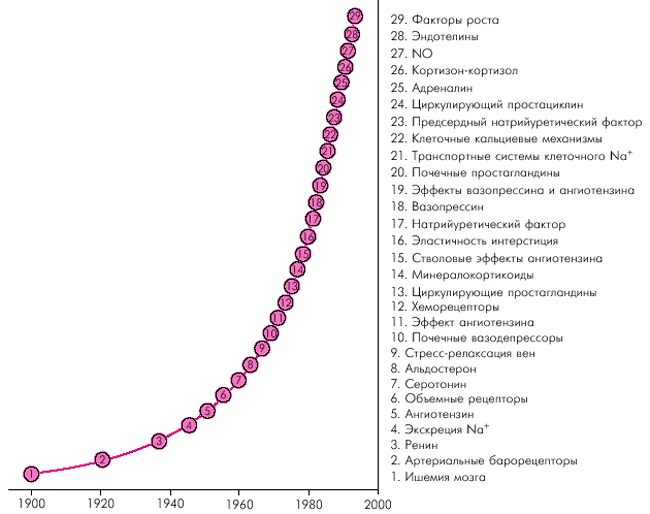
|
| Рис. 7.2. Наиболее значимые механизмы регуляции АД, изучавшиеся в ХХ веке (по Dickinson, в модификации) |
Нейрогенная концепция формированияАГ сложилась в 30–40-е годы прошлого столетия. Сторонники этой концепции (Г.Ф. Ланг, А.Л. Мясников и др.) придавали ведущее значение в патогенезе ГБ нарушениям центральной регуляции кровообращения, возникающим в результате “невроза” высших корковых и гипоталамических центров, который формируется под действием длительной психической травматизации и отрицательных эмоций. Эта гипотеза господствовала, как известно, в отечественной медицинской науке в течение нескольких десятков лет. Она дополнялась представлениями о нарушении при ГБ афферентного и эфферентного звеньев центральной регуляции — прессорных и депрессорных барорецепторов аорты и синокаротидной зоны, а также о гиперактивации САС.
Не отрицая значения нарушений высшей нервной деятельности в формировании гипертензивных реакций у больных АГ, роль “кардиоваскулярного невроза” в качестве пускового механизма возникновения ГБ все же представляется весьма сомнительной (Е.Е. Гогин, 1997). По современным представлениям, большее значение в формировании АГ имеют нарушения функционирования других механизмов регуляции АД: САС, РАС, РААС, калликреин-кининовой системы, ПНУФ, эндотелиальная дисфункция и т.д.
Роль гиперактивации симпато-адреналовой системы (САС). В большинстве случаев АГ, особенно на ранних стадиях формирования заболевания, протекает с выраженной гиперактивацией САС — гиперсимпатикотонией, которая является не столько результатом “кардиоваскулярного невроза” сосудодвигательного центра, сколько отражает дезадаптацию самой системы кровообращения к обычным физиологическим нагрузкам (физическим и эмоциональным).
Именно гиперсимпатикотония инициирует целый каскад регуляторных нарушений, так или иначе влияющих на уровень АД:
- увеличение сократимости ЛЖ и ЧСС, что сопровождается ростом сердечного выброса (МО);
- стимуляция норадреналином, выделяющимся в пресинаптической щели, α1-адренорецепторов гладкомышечных клеток артериол, что ведет к повышению сосудистого тонуса и величины ОПСС (рис. 7.3);
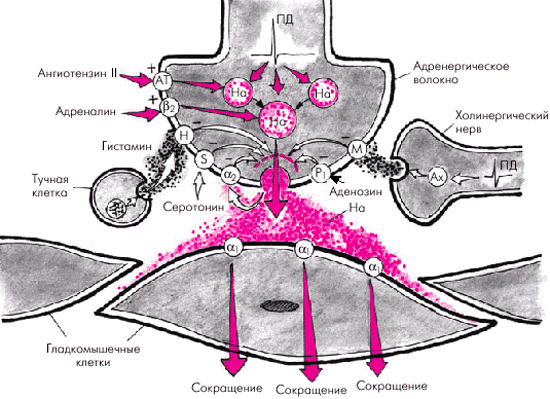
|
| Рис. 7.3. Повышение сосудистого тонуса, обусловленное стимуляцией aльфа1-адренорецепторов гладкомышечных клеток при гиперактивации САС. Красными стрелками обозначены вазоконстрикторные механизмы, белыми - механизмы, ограничивающие выделение норадреналина. На - норадреналин; АТ - рецепторы к ангиотензину; бета2 - бета2-адренорецепторы; Н - рецепторы к гистамину; S - рецепторы к серотонину; Р1 - рецепторы к аденозину; М - мускариновые рецепторы |
- стимуляция (через β-адренорецепторы) юкстагломерулярного аппарата почек (ЮГА), что приводит к активации РААС: ангиотензин II способствует повышению тонуса артериальной стенки, а альдостерон — задержке натрия и увеличению ОЦК.
- веноконстрикция, возникающая под действием норадреналина, ведет к увеличению венозного возврата крови к сердцу, возрастанию преднагрузки и МО.
Таким образом, на фоне гиперактивации САС повышается активность целого ряда прессорных механизмов, регулирующих АД: увеличивается МО, ОПСС, ОЦК и т.д.
Активация ренин-ангиотензин-альдостероновой системы (РААС). Активация РААС играет ведущую роль в формировании АГ и ее последствий, в частности гипертрофии миокарда ЛЖ и клеток гладкой мускулатуры сосудистой стенки. Усиление секреции ренина в ЮГА почек происходит, как известно, не только в результате падения перфузионного давления в сосудах почек, но и под действием усиленной симпатической импульсации, характерной для больных с формирующейся АГ. Под действием ренина, циркулирующего в крови, образуется ангиотензин I (АI), который, подвергаясь воздействию АПФ (преимущественно в легких, плазме и почках), превращается в ангиотензин II (АII) — главный компонент РАС.
В главах 1 и 2 подробно рассматривались основные эффекты активации этой системы. Напомним, что под действием основного компонента этой системы (ангиотензина II) происходит:
- системное повышение тонуса артерий мышечного типа и увеличение ОПСС;
- повышение тонуса вен и увеличение венозного возврата крови к сердцу, возрастание преднагрузки;
- положительный инотропный эффект, сопровождающийся увеличением сердечного выброса;
- стимуляция альдостерона и задержка Nа+ и воды в организме, в результате чего возрастает ОЦК и содержание Nа+ в гладкoмышечных клетках;
- стимуляция пролиферации кардиомиоцитов и гладкой мускулатуры сосудов.
Действие ангиотензина II на гладкомышечные клетки сосудов и кардиомиоциты опосредуется с помощью ангиотензиновых рецепторов — АТ1 и АТ2. Рецепторы АТ1 реализуют в основном вазоконстрикторные эффекты ангиотензина II, а рецепторы АТ2 — преимущественно стимуляцию клеточной пролиферации.
Следует помнить, что трансформация АI в АII может происходит не только под действием ангиотензинпревращающего фермента (АПФ). Возможен альтернативный путь образования АII с помощью тканевой химазы и других соединений.
Важно помнить, что РААС функционирует не только как эндокринно-гуморальная система, эффект которой обусловлен циркулирующим АII. Последний обеспечивает, главным образом, краткосрочные эффекты системной и регионарной циркуляции:
- системную и почечную вазоконстрикцию;
- усиление секреции альдостерона, реабсорбции Nа+ и воды почками;
- положительное хронотропное и инотропное влияние на миокард.
Эти влияния, несомненно, имеют большое значение в генезе АГ.
Еще более важным для формирования эссенциальной АГ имеет тканевой ренин- ангиотензиновый эндотелийзависимый механизм, регулирующий регионарное кровообращение различных сосудистых областей. Ангиотензин II, образующийся в тканях (в эндотелии сосудов), регулирует долговременные клеточные и органные эффекты РААС:
- местную и органную вазоконстрикцию, ведущую, в частности, к росту ОПСС;
- гипертрофию сосудистой стенки и миокарда ЛЖ;
- активацию фибропластического процесса в сосудистой стенке;
- активацию тромбоцитов;
- повышение тонуса эфферентных артериол клубочков и увеличение реабсорбции Nа+ в канальцах.
Тканевая РААС находится в тесном взаимодействии с другими эндотелийзависимыми факторами, как прессорными, так и депрессорными, оказывая существенное влияние на секрецию эндотелиального брадикинина, NО, эндотелинов и др.
Роль минералкортикоидов.Альдостерон и другие минералкортикоиды, вырабатываемые корой надпочечников (дезоксикортикостерон — ДОК и кортикостерон), обусловливают усиленную реабсорбцию Nа+ канальцами почек и ведут к задержке ионов Nа+ в организме. Избыток Nа+ способствует, в свою очередь, увеличению секреции вазопрессина — антидиуретического гормона (АДГ), что сопровождается уменьшением диуреза и задержкой воды в организме. Следствием этих двух процессов, как указывалось выше, является:
- увеличение ОЦК, ведущее, в том числе, к возрастанию АД;
- увеличение внутриклеточной концентрации ионов Nа+, а вслед за ними — ионов Са2+ (в соответствии с Nа+-Са2+–обменным механизмом), что резко повышает чувствительность сосудистой стенки даже к обычным физиологическим прессорным стимулам (катехоламинам и ангиотензину II);
- повышение внутриклеточной концентрации Nа+, способствующее набуханию и снижению эластичности сосудистой стенки, вследствие чего способность артерий расширяться во время прихода в данную сосудистую область пульсовой волны резко уменьшается.
Роль предсердного натрийуретического фактора (ПНУФ).Как известно, предсердный натрийуретический фактор (ПНУФ) принимает участие в сохранении нормального объема внеклеточной жидкости за счет стимуляции натрийуреза. Если имеется нарушение выделения почками ионов Nа+, которое сопровождается увеличением ОЦК и объема предсердий и желудочков сердца, активность ПНУФ и натрийурез возрастают. Обычно этот механизм реализуется за счет ингибирования предсердным натрийуретическим фактором клеточной Nа+-К+–АТФазы. В результате возрастает внутриклеточная концентрация Nа+ и, соответственно, ионов Са2+, что повышает тонус и реактивность сосудистой стенки.
Нарушение транспорта катионов через клеточную мембрану.В последние годы было показано (Ю.В. Постнов), что у больных эссенциальной АГ наблюдается значительное повышение проницаемости мембран для одновалентных ионов (Nа+, Са2+, Li+ и др.), что приводит к увеличению внутриклеточной концентрации ионов Nа+ и Са2+. Этому способствует также уменьшение связывания внутриклеточного Са2+ и выведения его из клетки. В результате возрастает внутриклеточная концентрация Са2+ и Nа+, а также тонус гладкой мускулатуры сосудистой стенки, и повышается ОПСС. Некоторые исследователи считают, что именно эти дефекты мембранного транспорта Са2+ и Nа+ лежат в основе наследственной предрасположенности к возникновению АГ (Ю.В. Постнов, В.Н. Орлов, Е.Е. Гогин и др.).
Нарушение экскреторной функции почек. Участие почек в патогенезе ГБ не ограничивается повышенным функционированием РААС или реализацией действия АДГ или ПНУФ. Большое значение, причем на самых ранних стадиях развития заболевания, имеют нарушения экскреторной функции почек, которые связывают с первичными наследственными дефектами внутрипочечной гемодинамики и ретенции Nа+ и воды почками. Характер таких дефектов не совсем ясен. J.H. Laragh (1989) и другие считают, что у больных эссенциальной АГ имеет место врожденный дефект части нефронов, который проявляется гипоперфузией этих нефронов, что в конечном счете приводит к закономерному возрастанию реабсорбции Nа+ в канальцах почек.
Согласно другой гипотезе, снижение экскреторной функции почек происходит в результате нарушения почечной гемодинамики, обусловленной первичным повышением тонуса выносящей артериолы почечных клубочков. В результате развивается внутриклубочковая гипертензия и гиперфункция нефронов, которая компенсируется усилением проксимальной реабсорбции.
Так или иначе, нарушение реабсорбции Nа+ и воды в почках признаются ведущими механизмами формирования эссенциальной АГ (ГБ) на всех этапах ее прогрессирования. В начальной стадии ГБ почки выполняют важные компенсаторные функции, направленные на поддержание достаточного натрийуреза и диуреза, а также снижение тонуса сосудистой стенки за счет активации почечных депрессорных систем (калликреин-кининовой системы и простагландинов). Со временем действие этих депрессорных механизмов становится недостаточным для поддержания нормального уровня АД. Мало того, в почках развиваются значительные структурные и функциональные изменения, при которых поддержание достаточного объема фильтрации и экскреции Nа+ и воды возможно только при условии сохранения высоких цифр АД. Таким образом, почка принимает участие в стабилизации АД на новом высоком уровне.
Ожирение и гиперинсулинемия. У части больных ГБ большое значение для формирования и прогрессирования АГ приобретают ожирение и характерные для него нарушения жирового, углеводного и инсулинового обменов. Как известно, клетки жировой ткани (адипоциты) существенно изменяют метаболизм и теряют чувствительность к обычным физиологическим стимулам — действию катехоламинов, ангиотензина, инсулина, симпатическим стимулам и т.д. В связи с этим у больных, страдающих ожирением, закономерно повышается активность САС, РААС, наблюдается гиперальдостеронизм, гипертрофируется кора надпочечников и т.п. В результате резистентности тканей к действию инсулина у больных ожирением, как правило, обнаруживают повышенный уровень инсулина (гиперинсулинемию), а также гипертриглицеридемию.
Как известно, гиперинсулинемия сопровождается:
- повышением активности САС;
- активацией РААС и задержкой Nа+ и воды в организме;
- стимуляцией развития гипертрофии сосудистой стенки.
Все три фактора являются важнейшими механизмами формирования и прогрессирования АГ. В последние годы много внимания уделяется изучению клинической картины и патогенеза так называемого “метаболического синдрома”, в основе которого лежит, как известно, наличие ожирения, инсулинорезистентности, гипертриглицеридемии и АГ. У лиц с метаболическим синдромом существенно повышен риск возниновения ИМ, внезапной сердечной смерти и сахарного диабета. В связи с этим N.M. Kaplan предложил называть сочетание таких факторов риска как ожирение, инсулинорезистентность, гипертриглицеридемия и АГ “смертельным квартетом”. Инсулинорезистентность и гиперинсулинемия рассматриваются в настоящее время в качестве пусковых факторов, инициирующих целый ряд механизмов, приводящих в конечном счете к развитию на фоне ожирения гиперлипидемии, АГ и ИБС.
Дисфункция эндотелия. Нарушениям функции эндотелия придается в настоящее время особое значение в формировании ряда распространенных заболеваний сердечно-сосудистой системы — атеросклероза, АГ, ИБС и сахарного диабета. Имеет значение продукция эндотелием NО, эндотелина, простациклина, цАМФ, брадикинина, тромбоцитарного активирующего фактора и ангиотензина II (тканевого).
Напомним, что в норме указанные соединения обеспечивают стабильность объема местного кровотока при колебаниях системного АД. Снижение АД ведет к повышению “секреции” депрессорных факторов (NО, простациклина, брадикинина, ЭГПФ и др.), компенсаторному расширению резистивных сосудов и поддержанию местного кровотока на должном уровне. Одновременно “включается” целый ряд прессорных систем, обеспечивающих восстановление системного АД (центральный аппарат регуляции АД, САС, РААС и т.п.).
Наоборот, в ответ на повышение системного АД усиливается продукция эндотелиальных прессорных соединений (эндотелины, тканевой АII, тромбоксан А2) и уменьшается “секреция” депрессорных субстанций. В результате происходит сужение местных резистивных сосудов и активное ограничение местного кровотока, что предотвращает избыточное поступление крови в жизненно важные органы и перегрузку его микроциркуляторного русла.
Как известно, повреждение эндотелия, обусловленное действием различных неблагоприятных факторов (гемодинамическая перегрузка, курение, алкоголь, возрастные инволютивные изменения эндотелия и др.), сопровождается нарушением его функционирования — дисфункцией эндотелия. Возникает неадекватный регуляторный ответ сосудистой стенки на обычные гемодинамические ситуации. У больных эссенциальной АГ обусловленная эндотелием вазодилатация подавляется за счет избыточной продукции субстанций, обладающих сосудосуживающим эффектом. При АГ особое значение приобретают активация тканевой эндотелийзависимой ренин-ангиотензиновой прессорной системы, избыточное выделение эндотелинов и угнетение тканевой калликреин-кининовой системы, оксида азота (NО), эндотелиального гиперполяризующего фактора (ЭГПФ) и т.д. (рис. 7.4).
| Рис. 7,4. Эндотелиальная дисфункция при гипертонической болезни с преобладанием вазоконстрикторных факторов и угнетением вазодилатирующих субстанций |
|
Следует иметь в виду тесную взаимосвязь метаболизма перечисленных эндотелиальных факторов (рис. 7.5). Поэтому активация тканевой РАС и ангиотензинпревращающего фермента (АПФ) не только способствует усиленной трансформации АI в АII по основному ферментативному пути, но и угнетает выработку основных депрессорных субстанций. Как известно, АПФ одновременно выполняет роль ключевого фермента калликреин-кининовой системы — кининазы II, которая быстро разрушает брадикинин. Последний обладает мощным вазодилатирующим эффектом, способствующим уменьшению тонуса гладкомышечных клеток сосудов. Кроме того, брадикинин, связываясь с В2-кининовыми рецепторами, усиливает образование других депрессорных субстанций: оксида азота (NО), простациклина (PGI2) и эндотелиального гиперполяризующего фактора (ЭГПФ). Поэтому увеличение активности АПФ сопровождается не только увеличением выработки тканевого АII, но и более быстрым разрушением брадикинина, что устраняет его стимулирующее влияние на выделение эндотелием NО, PGI2 и ЭГПФ. Одновременно увеличивается образование эндотелина, повышающего концентрацию внутриклеточного Са2+. В результате начинает преобладать эндотелийзависимая вазоконстрикция.
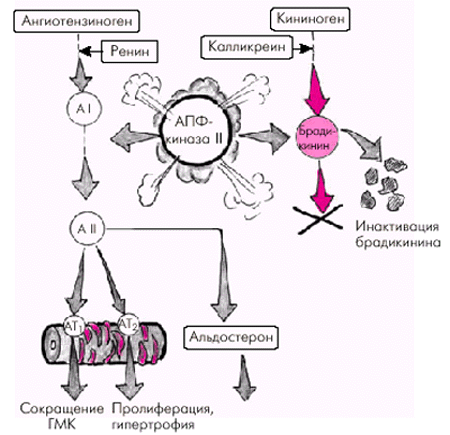
|
| Рис. 7.5. Роль ангиотензинпревращающего фермента (АПФ) в повышении сосудистого тонуса и ремоделировании сосудов при гипертонической болезни Роль ангиотензинпревращающего фермента (АПФ) в повышении сосудистого тонуса и ремоделировании сосудов при гипертонической болезни |
Таким образом, аномальное функционирование сосудистого эндотелия является одним из ведущих патогенетических звеньев развития эссенциальной АГ (ГБ).
Структурные изменения сосудистой стенки. Важнейшим фактором стабилизации повышенного АД являются структурные изменения сосудистой стенки, закономерно развивающиеся у больных ГБ вслед за функциональными нарушениями эндотелия. Возникает диффузная распространенная гипертрофия сосудистой стенки, возникающая, прежде всего, в результате активации местной тканевой РАС. Ангиотензин II, образующийся в избыточном количестве в эндотелии, воздействуя на ангиотензиновые рецепторы АТ2, приводит к пролиферации гладкомышечных клеток, частичному повреждению внутренней мембраны. Стенка артериол утолщается, средние и мелкие сосуды превращаются в жесткие трубки с узким просветом, неспособные расширяться.
Дата добавления: 2016-01-20; просмотров: 703;