Нарушение переваривания и всасывания липидов
Для нормального переваривания и всасывания их в кишечнике определяющее значение имеет взаимодействие таких факторов, как:
1) выработка поджелудочной железой липолитического фермента липазы;
2) поступление с желчью желчных кислот, эмульгирующих жиры и продукты их распада, активирующих панкреатическую липазу и участвующих во всасывании жирных кислот (всасывается комплекс жирных и желчных кислот);
З) захват продуктов переваривания липидов клетками слизистой оболочки тонкого кишечника;
4) превращение в стенке кишечника всосавшихся продуктов гидролиза липидов в частицы (хиломикроны) для дальнейшего транспорта их в лимфатические сосуды и далее в кровоток.
При нарушении любого из этих процессов развивается стеаторея - избыточное содержание жира в испражнениях.
Причинами нарушения переваривания и всасывания липидов являются:
1. Дефицит или низкая активность панкреатической липазы (поражение поджелудочной железы), что приводит к нарушению расщепления жиров.
2. Недостаточное поступление желчных кислот в кишечник (при гепатитах, циррозах, холециститах, обтурационной желтухе и др.) вызывает нарушение эмульгирования и расщепления жира, а также переноса продуктов его гидролиза к всасывающей поверхности эпителия кишечника.
3. Дефицит гормонов желудочно-кишечного тракта (холецистокинин, гастрин и др.), регулирующих сокращение стенок желчного пузыря, процессы эмульгирования и расщепления жиров, их транспорт через кишечную стенку.
4. Поражение эпителия тонкого кишечника различными ядами (флоридзин, монойодуксусная кислота) и инфекционными агентами, инактивирующими ферментные системы ресинтеза триацилглицеридов эпителия тонкого кишечника, а также процессы фосфорилирования и дефосфорилирования в стенке кишечника.
5. Авитаминозы А, В, С.
6. Избыточное потребление с пищей ионов Са2+ и Мg2+, что приводит к образованию нерастворимых в воде солей жирных кислот (мыла).
7. Дефицит холина в пище или недостаточное его образование из метионина при малобелковом питании тормозит реабсорбцию липидов.
8. Изменение деятельности нервной и эндокринной систем: перерезка блуждающего нерва ослабляет всасывание жиров из кишечника, аналогично действует наркоз; АКТГ и тироксин усиливают всасывание жира. При недостатке гормонов коры надпочечников или избытке адреналина всасывание жира замедляется.
9. Усиленная перистальтика кишечника и диарея препятствуют реабсорбции большей части жира.
10. Нарушение метаболизма липидов в энтероцитах с образованием аномальных белковолипидных комплексов ухудшает всасывание жира и вызывает образование жировых скоплений в стенке тонкого кишечника и в мелких лимфатических протоках, что блокирует отток лимфы.
Дефицит липидов в организме может быть связан не только с нарушением их всасывания в кишечнике, но и с усилением их выведения. Организм может терять липиды с мочой (липидурия), что наблюдается при липоидном нефрозе. Возможны потеря липидов сальными железами (экзема, угревая сыпь) и выход липидов из депо при травматизации больших участков жировой ткани и костного мозга.
Недостаток липидов в организме может привести:
1) к развитию гиповитаминозов (снижение содержания жирорастворимых витаминов А, D, Е, К);
2) к возникновению дефицита незаменимых полиненасыщенных жирных кислот с последующим нарушением синтеза биологически активных веществ (лейкотриены, простагландины и др.). Это, как правило, сопровождается выпадением волос, воспалительным поражением кожи, возникновением некротических очагов и экзематозных явлений, поражением почек, потерей способности к размножению;
З) к развитию истощения.
Гиперлипемия
Гиперлипемия является одним из показателей нарушения жирового обмена и характеризуется увеличением содержания липидов в крови.
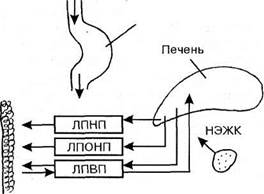
Липиды поступают в лимфу, а затем в кровь из кишечника в виде самых крупных липопротеидов - хиломикронов, из печени в кровь выходят липопротеиды очень низкой плотности. При липолизе из подкожной жировой клетчатки, легких, костного мозга освобождаются неэтерифицированные жирные кислоты (НЭЖК).
Уровень липидов в плазме крови в норме не превышает 1-2 г/л.
Гиперлипемия может быть алиментарной, транспортной и ретенционной.
Алиментарная гиперлипемия - временное увеличение уровня хиломикронов в крови, вызванное приемом жирной пищи или проведением пробы с липидной нагрузкой. Она легко устраняется с помощью возросшей функциональной активности гепатоцитов, утилизирующих хиломикроны. Возможно также усиление депонирования липидов в жировой ткани (рис. 89).

|
Рис. 89. Причины гиперлипемии (схема):
1 - усиленное поступление в кровь хиломикронов и жирных кислот из кишечника; 2 - усиленное поступление в кровь липопротеидов из печени; 3 - усиленное поступление в кровь НЭЖК из жировой ткани; 4 - низкая активность липопротеидлипазы; 5-6 - задержка поступления жирных кислот из крови в жировую ткань и мышцы; 7 - усиленное расщепление комплекса альбумина с жирной кислотой; 8 - гипоальбуминемия и недостаточное образование комплекса альбумина с жирной кислотой.
Заштрихованные столбики - место нарушения процесса
Транспортная гиперлипемия обусловлена либо усиленной мобилизацией из депо в виде неэтерифицированных жирных кислот при голодании, стрессе, сахарном диабете, либо нарушением метаболизма циркулирующих в крови липопротеидов при различных формах семейной гиперлипемии. Усилению мобилизации липидов из жировой ткани, костного мозга способствуют соматотропный и кортикотропный гормоны гипофиза, а также глюкагон, тироксин и адреналин, которые активируют тканевую липазу через аденилатциклазную систему.
Из печени липопротеиды (комплекс липидов с белками) поступают в кровь. Сами липиды гидрофобны и поэтому не образуют суспензии в плазме крови. Гидрофильность им обеспечивают белки.
Мобилизация жира из легких, приводящая к гиперлипемии, возникает также при длительной гипервентиляции легких, например у пловцов и профессиональных певцов.
Ретенционная гиперлипемия (от лат. retention - задерживать) развивается в результате задержки перехода нейтральных жиров из крови в
ткани. Возникает при атеросклерозе, ишемической болезни сердца, нефрозе, сахарном диабете, при механической желтухе, поступлении большого количества NaCl (ингибирует липопротеиновую липазу). В патогенезе этого вида гиперлипемии большое значение имеют следующие факторы:
1.Снижение уровня гепарина, активирующего фактор просветления (липопротеиновая липаза), - при нефрозе, механической желтухе, атеросклерозе.
2. Уменьшение содержания альбуминов в крови (осуществляют транспорт НЭЖК в клетки различных органов) - при нефротическом синдроме, заболеваниях печени и др.
3. Присутствие в сыворотке ингибитора липопротеиновой липазы - при нефротическом синдроме.
4. Снижение активности липокаина, активирующего поступление в кровь липопротеиновой липазы - при сахарном диабете.
11.5.3. Нарушение обмена липопротеидов (гиперлипопротеидемии и дислипопротеидемии)
Всосавшиеся в кровь неполярные липидные молекулы циркулируют в крови и лимфе в комплексе с полярными соединениями (белками). Существует большой спектр частиц, несколько отличающихся по размерам, плотности и составу. Среди них выделены 4 основные группы липопротеидов (ЛП).
1. Липопротеиды высокой плотности (ЛПВП, или α-ЛП). В состав ЛПВП входят 40-55% белка (процент от общей массы частицы), 27-30% фосфолипидов, 3-8% триглицеридов, 2-3% свободного холестерина, 14-20% эфиров холестерина. Они синтезируются паренхимой печени, в стенке тонкого кишечника и всегда присутствуют в плазме крови здоровых людей. Выполняют транспортную функцию, переводя избыток холестерина с поверхности сосудов в печень и выводя его излишек из клеток эндотелия (рис. 90).
 | |||
 | |||
|
| Хиломикроны |
| Желудок |
| Жировая клетка |
Эндотелий сосуда
Рис. 90. Обмен липидов (схема)
2. Липопротеиды очень низкой плотности (ЛПОНП, или пре-β-ЛП). Представляют очень неоднородный класс частиц с различным содержанием компонентов: 8-12% - белок, 10-12% - свободный холестерин, 18-20% -фосфолипиды, 3-6% - эфиры холестерина, около 50% - триглицериды. Они образуются в основном в гепатоцитах и в меньшем количестве - в слизистой кишечника, являются главной транспортной формой эндогенных триглицеридов. В плазме крови происходит трансформация ЛПОНП в β-ЛП (при участии ферментов липопротеидлипазы и лецитин-холестеринацилтрансферазы - ЛХАТ крови). В ходе их катаболизма размеры частиц уменьшаются, меняется их состав (теряются триглицериды и возрастает относительный процент холестерина).
3. Липопротеиды низкой плотности (ЛПНП, или β-ЛП) имеют следующий состав: 24-31% - свободный холестерин, 16-28% - этерифицированный холестерин, 7-11% - триглицериды, около 30% - фосфолипиды, 20-25% - белок. Они образуются в плазме из ЛПОНП и являются самой атерогенной фракцией липопротеидов у человека.
4. Хиломикроны (ХМ) - самые крупные липопротеидные частицы, поступающие в кровь из лимфы и представляющие собой транспортную форму пищевых жиров (экзогенных триглицеридов). В их составе находятся: 3-8% фосфолипидов, 2-4% эфиров холестерина, около 2% свободного холестерина, 1-2% белка и 86-94% триглицеридов. Хиломикроны образуются в стенке кишечника в процессе всасывания экзогенных триглицеридов и холестерина, проникают в лимфу, а оттуда в кровеносные сосуды. В плазме крови они расщепляются под действием липопротеидлипазы и теряют значительное количество триглицеридов (образуются СЖК и глицерин). Для ткани легких катаболизм ХМ особенно важен, поскольку играет ключевую роль в обеспечении высокой активности альвеолярных макрофагов и необходим для синтеза фосфолипидов сурфактанта (рис.91).

Рис. 91. Роль сурфактанта в предупреждении ателектаза
В связи с этим при заболеваниях легких положительный эффект дает жировая диета. Следует отметить, что плазма крови здоровых людей натощак (через 12-14 ч после приема пищи) не содержит ХМ.
При ряде заболеваний липопротеидный спектр сыворотки меняется и возникают гипер- или гипо-(а)липопротеидемии. При этом наблюдаются увеличение или, наоборот, снижение содержания, вплоть до полного отсутствия одного или нескольких классов липопротеидов в крови, а также появление их определенных форм (дислипопротеидемии).
Различают 5 типов гиперлипопротеидемий (ГЛП):
I. Гиперхиломикронемия - характеризуется высоким содержанием хиломикронов в плазме натощак. Проявляется ксантоматозом - это отложение холестерина и его эфиров в купферовских клетках печени, гистиоцитах подкожной клетчатки и сухожилиях с последующим разрастанием соединительной ткани в виде бляшек и узлов желтоватого цвета.
У больных развивается гепатоспленомегалия, наблюдаются тромбоз и микронекрозы поджелудочной железы с последующим формированием хронического панкреатита, абдоминальные колики после принятия жирной пищи. На коже видны ксантомы в виде желтоватых папул. Заболевание может быть вызвано наследственным аутосомно-рецессивным дефектом липопротеиновой липазы либо аутоиммунными заболеваниями соединительной ткани (при системной красной волчанке образуются антитела против гликозаминогликанов, что нарушает процесс гепариновой активации липопротеидлипазы).
II. Гипер-β-липопротеидемия делится на 2 типа:
IIа - увеличение содержания в крови β-ЛП при нормальном уровне пре-β-ЛП
IIб - увеличение содержания β-ЛП и пре-β-ЛП.
Для заболевания характерен выраженный ксантоматоз век, кожи, роговицы, развитие ишемической болезни сердца с инфарктом миокарда в очень раннем возрасте, атеросклеротические поражения сосудов у детей. Предполагается, что в основе заболевания лежит аутосомно-доминантный дефект рецепторов ЛПНП (IIа), либо нарушение взаимодействия рецепторов на клеточных мембранах с ЛПОНП и ЛПНП, либо дефект липопротеидлипазы (IIб).
III. «Флотирующая» гиперлипопротеидемия, или дис-β-липопротеидемия. В основе заболевания лежит наследственно обусловленное нарушение синтеза апопротеина Е (белок, входящий в состав ХМ и ЛПОНП). Заболевание характеризуется появлением в сыворотке флотирующих β-ЛП, которые называются промежуточными. Они обогащены холестерином, а содержание триглицеридов в них может быть снижено. Образуются эти частицы при нарушении катаболизма ЛПОНП и ХМ. Встречаются также приобретенные формы заболевания при гипотиреозе, танжерской болезни, некоторых аутоиммунных гаммапатиях.
Этот вид ГЛП сопровождается ранними атеросклеротическими проявлениями (после 20 лет), развитием ИБС, ишемической энцефалопатии вплоть до инсультов, ксантоматозом, ожирением.
IV. Гипер-пре-β-липопротеидемия. Заболевание может быть наследственно обусловленным (аутосомно-доминантное) или приобретенным (при алкоголизме, остром гепатите, акромегалии, диабете и др.). Патогенез до конца не выяснен. Для этого типа ГЛП характерно нарастание уровня триглицеридов и ЛПОНП в крови. Содержание ЛПНП и ЛПВП варьирует от нормального до значительно сниженного. У больных развиваются ожирение и сахарный диабет, появляются ксантомы, возможны атеросклеротическое поражение сосудов нижних конечностей, липидоз сетчатки и ухудшение зрения, проявления ишемической болезни сердца.
V. Гипер-пре-β-липопротеидемия и хиломикронемия. При этом заболевании в крови увеличивается содержание ХМ и ЛПОНП и снижается уровень ЛПНП и ЛПВП. У больных отмечаются гепато- и спленомегалия, ожирение, снижение толерантности к глюкозе, поражение миокарда. После приема жирной пищи могут наблюдаться внезапные приступы абдоминальной колики, ксантоматоз и атеросклероз слабо выражены
В патогенезе первичного заболевания главную роль играет наследственно обусловленное отсутствие кофактора липопротеидлипазы - апопротеина СП (аутосомно-рецессивное наследование), в результате два основных субстрата воздействия этого фермента накапливаются в крови.
Фенокопия болезни развивается при алкоголизме, гликогенозе Гирке и некоторых других заболеваниях печени.
Гипо-(α)липопротеидемии (относительно редкие аномалии спектра ЛП)
1. А-β-липопротеидемии В основе заболевания лежит аутосомно-доминантный дефект синтеза апопротеина В (белковой части липопротеидов), что приводит к аномалии строения хиломикронов, снижению содержания или полному отсутствию в плазме ЛПОНП и ЛПНП. Клинические проявления связаны с нарушением всасывания в кишечнике жиров и углеводов, гемолитической анемией, дегенерацией бокового и заднего канатиков спинного мозга, пигментной ретинопатией. Нарушение всасывания жиров проявляется сразу после рождения плохим аппетитом, рвотой, обильными испражнениями, стеатореей, развитием гипотрофии. Примерно у трети больных развивается умственная отсталость. С возрастом усиливаются неврологические расстройства, появляются скелетные деформации, сердечные аритмии, ухудшается зрение. В патогенезе заболевания решающее значение имеет снижение содержания холестерина в клеточных мембранах и потеря жирорастворимых витаминов, особенно витамина Е, что ведет к утрате антиоксидантной защиты мембран.
2. Танжерская (тэнжирская) болезнь. В основе заболевания лежит аутосомно-рецессивное нарушение синтеза апопротеина А, что, в свою очередь, нарушает продукцию ЛПВП. У больных нарушен транспорт эфиров холестерина, в результате эфиры захватываются макрофагами и откладываются в клетках ретикуло-эндотелиальной системы селезенки, печени, лимфоидных органов. Наблюдается лимфаденопатия, гепатоспленомегалия, неврологические нарушения - слабость, парестезии, снижение сухожильных рефлексов. Одним из ярких признаков заболевания является оранжево-желтый цвет увеличенных миндалин.
Существуют и другие формы гиполипопротеидемий: церебросухожильный ксантоматоз (наследственный дефект синтеза желчных кислот из холестерина), болезнь Вальмана (аутосомно-рецессивный дефицит холинэстеразы), гипоальфалипопротеинемия (генетически детерминированное нарушение продукции апопротеина А и С) и др. Большинство из них связано с наследственной патологией синтеза белковой части липопротеидов либо с нарушением метаболизма холестерина.
Дата добавления: 2016-01-07; просмотров: 5236;