Особенности химии переходных элементов.
Переходные элементы. Переходными называют элементы, в атомах или ионах которых d- и f-оболочки частично заполнены электронами. Так, атом серебра имеет электронную конфигурацию [Kr]4d105s1, то есть его d-подуровень полностью завершен, и в то же время в определенных условиях он обладает способностью отдавать d-электроны, например, переходя в степень окисления +2 с конфигурацией [Kr]4d9, известную в некоторых комплексных соединениях. Таким образом, серебро является переходным элементом. Элементы следующей группы, цинк, кадмий и ртуть с конфигурацией (n – 1)d10ns2, не образуют устойчивых соединений в степени окисления выше +2, то есть не теряют электроны с d-подуровня и, таким образом, не относятся к переходным. В то же время, наличие низкого по энергии целиком заполненного d-подуровня делает их химию во многом близкой химии переходных элементов, что позволяет рассматривать их в томе, посвященном переходным металлам.
В коротком варианте Периодической системы переходными считаются элементы побочных подгрупп (за исключением подгруппы цинка), а в длинном варианте - это элементы 3 – 11 групп.
Все переходные элементы характеризуются относительно низкими первыми потенциалами ионизации, что присуще металлам. По характеру заполняемых орбиталей переходные элементы подразделяются на d-элементы и f-элементы. d-Элементы образуют три ряда – в четвертом (3d), пятом (4d) и шестом (5d) периодах. f-Элементы объединяют в два семейства – лантаноиды (4f) и актиноиды (5f).
Согласно принципу Паули и правилам Клечковского, 3d- и 4d-подуровни заполняются сразу после 4s- и 5s-подуровней, соответственно. Поэтому d-элементы первых двух переходных рядов (3d и 4d) следуют непосредственно за элементами s-блока. В третьем переходном ряду у лантана также начинает заполняться d-подуровень (5d16s2), однако затем энергетически более устойчивым оказывается 4f-подуровень, энергия которого резко понижается при возрастании Z от 57 до 60. У церия (Z=58) на 5d-оболочке еще сохраняется единственный d-электрон (4f15d16s2), однако уже у следующего за ним празеодима (Z = 59) он переходит на 4f-подуровень (4f36s2). Далее вплоть до иттербия происходит заполнение 4f-подуровня, а у последнего из лантанидов – лютеция электрон поступает на 5d-орбиталь (4f145d16s2). Таким образом, лантан (Z = 57) и лютеций (Z = 71) формально считаются d-элементами. В то же время, по многим свойствам они близки 4f-элементам и рассматриваются вместе с ними. Все эти элементы, а также скандий и иттрий, часто объединяют в семейство редкоземельных. У следующего за лютецием элемента гафния (Z = 72) продолжается заполнение 5d-подуровня. Химия 5d-элементов во многом имеет большее сходство с химией металлов 4d-ряда, чем химия 3d- и 4d-металлов.
Частично заполненные d-орбитали активно участвуют в образовании химических связей. Такие связи, как правило, имеют преимущественно ковалентный характер. Степень ковалентности резко возрастает при уменьшении числа d-электронов, то есть, с повышением степени окисления, что сказывается в изменении температур плавления и кипения. Например, оксид марганца (II) плавится при 1840 °C, оксид марганца (II, III) Mn3O4 – при 1564 °C, а марганцевый ангидрид Mn2O7 – при 5 – 9 °C.
Элементы-лантаниды во многом отличаются от элементов d-блока. Внутренние 4f-орбитали сильно экранированы и практически не участвуют в образовании химических связей. Именно поэтому многие соединения лантанидов являются ионными, а в соединениях они стремятся иметь степень окисления +3. Исключение составляет церий, первый в ряду лантанидов. Для него характерна энергетическая близость 4f- и 5d-подуровней, что делает химию церия похожей на химию d-элементов. Аналогичная ситуация наблюдается и у элементов-актинидов вплоть до америция, эти элементы также похожи на d-переходные. Им свойственны переменные степени окисления, высокая доля ковалентности в соединениях. Актиниды конца 5f-ряда по свойствам напоминают лантаниды. Однако сильная радиоактивность этих элементов серьезно препятствует изучению их химии.
В отличие от непереходных элементов, в атомах металлов d-блока существенную роль играют d-электроны. Они расположены достаточно близко от ядра (Рис. 1.1. Зависимость радиальных волновых функций 3d-, 4s-, 4p-орбиталей атома кобальта от расстояния) и оказывают существенное экранирование, степень которого возрастает с заселением d-подуровня. При движении вдоль d-ряда, т.е. при последовательном переходе от элементов 3 группы к элементам 12 группы, наблюдаются рост эффективного заряда ядра, увеличение устойчивости d-подуровня и сжатие d-орбиталей (d-сжатие), сопровождающееся понижением энергии орбиталей (Рис. 1.2. Зависимость энергии атомных орбиталей от порядкового номера элемента) и уменьшением атомного радиуса. У элементов 12 группы энергия d-электронов оказывается ниже энергии внутренних орбиталей (рис. 1.3. Схематическое изображение изменения энергии d-, s-, p-орбиталей по ряду переходных элементов), иными словами, находящиеся на них электроны уже не являются валентными.
Электронная конфигурация. Практически все d-переходные элементы имеют в основном состоянии конфигурацию (n – 1) dynsx, где x = 1 – 2, а y = 1 – 10, то есть содержат хотя бы один s-электрон. Из-за более резкого уменьшения энергии d-орбиталей при движении по d-ряду и гораздо большего их сжатия по сравнению с s-орбиталями d-электроны испытывают более сильное отталкивание, что и вызывает расположение части электронов на s-орбитали, несмотря на то, что она обладает большей энергией (Рис. 1.2, 1.3.). Известно, что электроны с параллельными спинами испытывают меньшее взаимное отталкивание, чем электроны с противоположными спинами, разница в энергиях этих двух состояний называется обменной энергией К. Она достигает максимума в случае половинной и полной заселенностей d-подуровня, то есть при электронных конфигурациях d5 и d10. С этим связано явление «проскока электрона», наблюдающееся в электронных конфигурациях основного состояния элементов середины и конца d-рядов. Так, например, у хрома электронная конфигурация оказывается 3d54s1 вместо3d44s2, у палладия 4d105s0 вместо 4d84s2, у элементов группы меди (n – 1)d10ns1 вместо (n – 1)d9ns2.
Из-за большей энергии внешнего s-подуровня по сравнению с d-подуровнем ионизация атомов переходных металлов сопровождается потерей s-электронов и лишь затем электронов с d-подуровня. Так, например, электронная конфигурация атома железа [Ar]3d64s2, иона Fe2+ [Ar]3d6, а иона Fe3+ [Ar]3d5.
Атомные радиусы. Небольшое уменьшение радиусов при движению по d-ряду является следствием d-сжатия, вызванного постепенным повышением заряда ядер и усилением кулоновского притяжения электронов к ядру. По группе радиус заметно возрастает при переходе от 3d- к 4d-элементам и лишь незначительно при переходе от 4d- к 5d-элементам. Близость радиусов элементов-аналогов второго и третьего переходных рядов связана с заполнением 4f-подуровня, который предшествует заселению 5d. Уменьшение атомного радиуса за счет сокращения размера внутренних f-орбиталей (лантанидное сжатие) практически полностью компенсирует его рост, связанный с появлением нового энергетического уровня. Это приводит к близости химии переходных элементов начала ряда – циркония и гафния, в меньшей степени ниобия и тантала. На величины атомных радиусов, а также энергии ионизации тяжелых (5d) переходных элементов оказывают влияние релятивистские эффекты.
Энергия ионизации. По сравнению с s- и p-элементами у переходных металлов не наблюдается существенных изменений в энергиях ионизации при движении по периоду или по группе. В каждом из трех семейств d-элементов (3d, 4d, 5d) с увеличением числа d-электронов первая и вторая энергии ионизации несколько повышаются, что связано с общим увеличением устойчивости d-подуровня (Рис. 1.4. Изменение первой (а) и второй (б) энергий ионизации у 3d-элементов). Резкое увеличение энергии ионизации наблюдается у частиц с конфигурациями d5 или d10 (например, Cr+, Cu+, Zn0), обладающими высокими значениями обменной энергии и повышенной стабильностью. По тем же причинам частицы, содержащие один или шесть d-электронов, имеют пониженные значения энергий ионизации по сравнению с соседними в ряду d-элементов (I(Fe2+) = 30,65 эВ, сравните с I(Mn2+) = 33,67 эВ и I(Co2+) = 33,50 эВ).
Электроотрицательность. Если в малых периодах, состоящих из непереходных элементов, происходит существенный рост электроотрицательности, то при движении по d-ряду электроотрицательность возрастает лишь незначительно, как это наблюдалось для энергий ионизации. В группе электроотрицательность несколько убывает, наиболее заметно при переходе от 3d- к 4d-элементам. Так как d-орбитали металла принимают активное участие в образовании связей, электроотрицательность атома в соединении зависит от типа окружения, а в комплексах – от типа лиганда и его донорно-акцепторных свойств. Поэтому значения электроотрицательностей, рассчитаннные для нейтрального атома, в случае переходных металлов не являются информативными.
Координационные числа. Для элементов 3d-ряда наиболее распространено координационное число 6, хотя известны также многочисленные комплексы с пониженными координационными числами – 2, 3, 4, 5. Комплексы с координационным числом 4 имеют геометрию плоского квадрата или тетраэдра (Рис. 1.5 а, б). Для координационного числа 5 характерна форма квадратной пирамиды или тригональной бипирамиды (Рис. 1.5., в, г). Иногда встречаются и более высокие координационные числа – вплоть до 8. Для 4d- и 5d-элементов, особенно для тех, которые расположены в начале ряда, характерны высокие координационные числа – от 7 до 12. Изменение координационного числа наблюдается даже в соединениях со сходной стехиометрией. Так, например, твердый TiBr4 образован тетраэдрическими молекулами, в то время как ZrBr4 и HfBr4 имеют слоистую структуру и состоят из октаэдров, объединенных ребрами. Аналогично, хромовый ангидрид CrO3 построен цепями тетраэдров, связанных вершинами, а молибденовый MoO3 и вольфрамовый WO3 ангидриды имеют слоистую структуру, составленную из искаженных октаэдров с общими вершинами. Большинство соединений с координационным числом 6 имеют октаэдрическую геометрию, которая при определенных условиях (эффект Яна-Теллера) может искажаться.
Существуют три типа искажений октаэдра (Рис. 1.6): (1) тетрагональное, то есть растяжение или сжатие октаэдра по оси четвертого порядка (за противоположные вершины, рис. 1.6.А), (2) тригональное, то есть растяжение по оси третьего порядка (за противоположные грани, рис. 1.6. Б), образующийся полиэдр называют тригональной антипризмой, (3) ромбическое – одновременное растяжение по оси четвертого порядка и сжатие по оси третьего (рис. 1.6. В). Координационное число 7 реализуется в виде трех типов полиэдров – пентагональной бипирамиды, одношапочной тригональной призмы (Рис. 1.5.д) и одношапочного октаэдра (Рис. 1.5 е). Для координационного числа 8 характерна геометрия в форме куба или квадратной (архимедовой) антипризмы, образующейся при повороте одной из граней куба на 45° (Рис. 1.5.ж). Многие комплексы с координационным числом 9 имеют геометрию трехшапочной тригональной призмы (Рис. 1.5. з). Для актинидов и лантанидов известны ионы и с более высокими координационными числами – вплоть до 16 (M. C. Favas, D. L. Kepert, Prog. Inorg. Chem., 1981, 28, 309).
$$$$$$$$$$$$$$$$$
Характер химической связи в соединениях. В большинстве случае во взаимодействии металл-лиганд атом металла благодаря наличию вакантных d-рбиталей выступает в роли акцептора, а лиганд – в роли донора. Активное участие d-орбиталей в химической связи придает ей частично ковалентный характер. Степень ковалентности возрастает с ростом степени окисления и уменьшением координационного числа. Общий характер изменения доли ковалентности иллюстрирует график, составленный по результатам анализа данных кристаллических структур карбоксилатов различных металлов (Рис. 1.7. Зависимость длины связи и доли ковалентности карбоксилатов переходных металлов от номера группы) (R. K. Hocking, T. W. Hambley, Inorg. Chem., 2003, 42, 2833).
В основе взаимодействий металл-лиганд лежит проявление d-металлом роли акцептора, и лигандом – роли σ-донора. Доля ковалентности возрастает также при увеличении кратности связи М-М, которая в некоторых случаях может достигать четырех. При образовании кратных связей металл может выступать как в роли донора, так и в роли акцептора электронов. Переходные металлы в низких степенях окисления предоставляют часть d-электронов на вакантные орбитали лиганда. Такое взаимодействие называют π-дативным. Оно имеет место в комплексах переходных металлов с СО, олефинами, алкадиенами, фосфинами, молекулярным водородом. π-Акцепторные свойства лигандов усиливаются в ряду: NO > PF3 > CO > PCl3 > P(OR)3 > PR3 (где R – алифатический заместитель). Эти лиганды выступают одновременно и как σ-доноры, что приводит к образованию прочной химической связи (Рис. 1.8. Образование σ- (а) и π-связей (б) в комплексе с PF3). Данные соединения, из которых наиболее известны карбонилы, подчиняются правилу Сиджвика (правило эффективного атомного номера), согласно которому устойчивой является восемнадцати-электронная оболочка – именно она соответствует устойчивой элеткронной конфигурации (n-1)d10ns2np6 атома инертного газа. Так, в соответствии с этим правилом, никель в степени окисления 0 (Ni 3d84s2, 10 электронов) образует карбонил Ni(CO)4 (каждая молекула СО предоставляет пару электронов), а железо (Fe 3d64s2, 8 электронов) – Fe(CO)5. В случае кобальта (Co, 3d74s2, 9 электронов) восемнадцати-электронная оболочка может быть достигнута только путем образования димера со связью металл-металл Co2(CO)8, что и подтверждается экспериментально.
Акцепторные свойства металла наиболее ярко выражены в случае высоких степеней окисления и проявляются в соединениях с сильными σ-донорами. σ-Донорные свойства лигандов усиливаются с ростом отрицательного заряда и уменьшением размера O2– >> F–, P3– > S2– >> Cl–; N3– >> P3–. Кратная связь М–О в анионах МО4n– (M = V, Cr, Mn, Fe, Mo, Tc, Ru, W, Re, Os и др.) реализуется в результате взаимодействия заполненных орбиталей кислорода с пустыми d-орбиталями металла.
В то время как компактные 3d-орбитали перекрываются с орбиталями других атомов лишь на расстоянии, существенно меньшем суммы ковалентных радиусов, 4d- и 5d-орбитали, благодаря гораздо большей диффузности (Рис. 1.9. Сравнение радиальных волновых функций 3d-, 4d-, 5d-орбиталей), взаимодействуют с орбиталями лигандов, даже если они находятся на расстоянии, примерно равном сумме ковалентных радиусов. Поэтому в соединениях тяжелых переходных элементов (4d, 5d) связи металл-лиганд прочнее, а ковалентность выше, чем в случае 3d-элементов. По этой же причине оказываются устойчивыми и соединения со связью металл-металл – кластеры.
Дополнение – Кластеры
Термин кластер в современной науке имеет необычайно широкое значение и фактически может быть применен к любым объектам, представляющим собой скопление частиц в соответствии с английским значением этого слова. Так, ассоциаты молекул воды, присутствующие в жидкой и газовой фазах, нередко называют кластерами воды и т.д. В узком смысле слова кластером называют частицу со связью металл-металл. Простейшими объектами такого типа являются ультрадисперсные металлические частицы с диамтером менее 0.3 мкм и содержащие менее миллиона атомов. По мере уменьшение размера частиц в таком гигантском кластере возрастает роль атомов, находящихся на поверхности, которые стремятся к ассоциации с другими подобными им частицами. Единственный способ стабилизации заключается в покрытии их поверхности лигандами – карбонилом СО, галогенидами, фосфинами и др. Такие молекулярные кластерные соединения металлов часто обладают высокой устойчивостью, могут быть перекристаллизованы из различных растворителей, очищены ваккумной сублимацией. В некоторых случаях отдельные кластерные частицы оказываются соединенными друг с другом связями металл-металл в цепи, слои и каркасы. Так возникает кластерный материал.
В кластерах переходных металлов химическая связь осуществляется путем обобществления d-электронов, которые могут принимать участие и в образовании связи металл-лиганд. Для простейших карбонильных кластеров действует сформулированной Сиджвиком правило эффективного атмоного номера, предполагающее, что все связи в кластере являются двухцентровыми двухэлектронными. Только в этом случае частица с 18 валентными электронами оказывается устойчивой. В многоядерных полиэдрических кластерах, в вершинах которых сходится не менее четырех ребер, приавило Сиджвика нарушается, что свидетельствует о более сложном характере связи, описываемом методом молекулярных орбиталей. Большинство галогенидных кластеров также не подчиняется правилу Сиджвика: эти соединения элеткронодефицитны, подобно боранам. Иными словами, валентных элеткронов не хватает на образование двухцентровых двухэлектронных связей. Стабильность таких соединений удается предсказать, подсчитав число кластерных скелетных электронов, то есть электронов, участвующих в образовании связи металл-металл. Оно равно общему числу валентных электронов металла за вычитом электронов, задействованных в образовании связи металл-лиганд и заряда кластерной частицы. Например, в Re3Cl9, оно равно 7×3 – 9 = 12, что соответствует трем четырехкратным связям металл-металл, а в Mo6Cl12 6×6 – 2 –12 = 24, то есть в октаэдре Мо6, имеющем 12 ребер реализуется 12 одинарных связей Мо-Мо. Уменьшение числа кластерных электронов приводит к ослаблению связей между атомами металла и в конечном счете делает образование кластерной частицы невыгодной. Часто кластерные частицы без изменения переходят из одного соединения в другое, в иных случаях изменяется лишь ее заряд. Конкретные типы кластерных соединений будут обсуждаться в главах, посвященных химии элементов (С. П. Губин, Химия кластеров, М., Наука, 1987; J.D. Corbett, Inorg. Chem., 2000, 39, 5178)
Рис. 1.10. Кластеры (а) InZn8–, (б) Mo6Cl86+, (в) Pr4RuI5
Степени окисления. d-Элементы в соединениях проявляют несколько степеней окисления, от низших, представленных преимущественно нейтральными или катионными формами (Mn2+, Mo(CO)6), до высших, существующих в форме анионов (MnO42– , ReO4–). Наибольшее разнообразие степеней окисления характерно для металлов середины каждого d-ряда (Рис. 1.11. Наивысшие известные степени окисления переходных элементов), а также для некоторых элементов-актинидов (урана, нептуния, плутония, америция). Наивысшая степень окисления (+8) известна для платиновых металлов восьмой группы – рутения и осмия. В противоположность p-элементам, устойчивость высших степеней окисления вниз по группе возрастает, что связано с увеличением размера d-орбиталей и сопутствующим усилением ковалентности. Таким образом, соединения 3d-металлов в низких степенях окисления преимущественно ионные, в высших степенях окисления – ковалентные. Например, из высших оксидов элементов 8 группы известны лишь RuO4 и OsO4, марганцевый ангидрид Mn2O7 разлагается при 55 °C со взрывом, в то время как Tc2O7 и Re2O7 плавятся без разложения выше 200 °C. При движении вниз по подгруппе восстановительные свойства простых веществ и соединений ослабевают, а окислительные усиливаются (Табл. 1.1).
Таблица 1.1
Стандартные электродные потенциалы некоторых d-элементов в высших степенях окисления.
| Процесс | E0, B (pH = 0) | Процесс | E0, B (pH = 14) | Процесс | E0, B (pH = 14) |
| MnO4–/MnO2 TcO4–/TcO2 ReO4-/ReO2 | 1,68 0,74 0,51 | CrO42–/Cr MoO42–/Mo WO42–/W | –0,80 –0,91 –1,07 | FeO42–/Fe2+ RuO42–/Ru2+ OsO42–/Os2+ | 1,84 1,56 0,99 |
Так, перманганат-ион MnO4– проявляет ярко выраженные окислительные свойства, в то время как для восстановления перрената ReO4– необходим сильный восстановитель. Для химии 4d- и 5d-элементов вообще характерны высокие степени окисления. Если в соединениях марганца степень окисления +2 преобладает, то для его тяжелых аналогов – технеция и рения – в этой степени окисления известно всего несколько простых соединений. Некоторые из соединений тяжелых переходных элементов (RuO4, WCl6, OsF7) вообще не имеют аналогов среди 3d-металлов.
Необходимо помнить, что степень окисления представляет собой условный заряд, который, как правило, значительно отличается от реального. Чем меньше размер катиона и чем выше заряд, тем менее вероятно его существование в водном растворе.
Низшие степени окисления переходных металлов устойчивы в соединениях с π-акцепторами – карбонилом, цианидом, олефинами, фосфинами. Примерами могут служить комплексы родия(+1) с олефинами (Rh2(C2H4)4Cl2), цианидный комплекс никеля в нулевой степени окисления Na4[Ni(CN)4], многочисленные карбонилы металлов, карбонильный комплекс железа(–2) [Fe(CO)4]2–. В водных растворах низшие степени окисления более устойчивы в кислой среде. Так, марганец(+2) в нейтральных и кислых растворах не взаимодействует с кислородом воздуха, в то время как в присутствии щелочи быстро окисляется. Так же ведут себя и многие другие переходные элементы.
Высшие степени окисления устойчивы в соединениях с наиболее электроотрицательными элементами – фтором (RhF6, K3NiF6, Cs2CuF6), кислородом (K2FeO4, OsO4) - сильными акцепторами электронов. Многие соединения в высоких степенях окисления получены в форме фторидов. Их окислительные свойства наиболее сильны в кислой среде (Сноска: о неустойчивости высших степеней окисления в кислой среде см. S.M. Elder, G.M. Lucier, F.J. Hollander, N. Bartlett, Journ. Amer. Chem. Soc., 1997, 119, 1020), поэтому синтез проводят в присутствии щелочных реагентов. Например, окисление гидроксида хрома(III) до хромата(VI) под действием брома, пероксида водорода или гипохлорита легко протекает в щелочной среде, но сильно затруднено в кислой.
Стабилизировать элемент в неустойчивой степени окисления – значит понизить его окислительную или восстановительную активность. Это можно сделать, изменив его электродный потенциал, например, переведя данный элемент в трудно растворимое соединение или в комплекс. Чем ниже произведение растворимости осадка или чем выше константа устойчивости комплекса, тем больше шансов на успех. Известно множество примеров подобных превращений. Марганец(III) неустойчив в водных растворах в форме аква-ионов, однако малорастворимый оксогидроксид MnOOH (существует как минерал) и оксалатный комплекс K3[Mn(C2O4)3] устойчивы в водной среде. Многие неустойчивые степени окисления удается стабилизировать в виде кислородных соединений (K3FeO2, BaFeO3, K3FeO4; смотри F. Bernhardt, R. Hoppe,, Z. Anorg. Allg. Chem., 1993, 619, 969) фторидных комплексов, гетерополисоединений, периодатов (Na4K[Cu(HIO6)2]×12H2O).
Магнитные свойства. Магнитные свойства атомов и ионов определяются наличием в них электронов. Электрон обладает как собственным (спиновым) магнитным моментом, обусловленным его вращением вокруг собственной оси, так и орбитальным, вызванным его движением вокруг ядра. В большинстве соединений 3d-переходных элементов в суммарный магнитный момент основной вклад вносит спиновая составляющая, которую рассчитывают по формуле
 , где n – число неспаренных электронов.
, где n – число неспаренных электронов.
Атомы и ионы, содержащие неспаренные электроны, называются парамагнитными. Парамагнитные частицы втягиваются в магнитное поле. Если спиновый магнитный момент частицы равен нулю, она является диамагнитной, то есть обладает лишь орбитальным магнитным моментом. Диамагнетики не содержат неспаренных электронов, а при попадании во внешнее магнитное поле выталкиваются из него.
Вещества по магнитным свойствам делятся на несколько классов. Помимо парамагнетиков (алюминий, натрий, хлорид меди(II)) и диамагнетиков (хлорид натрия, аргон), известны ферро- и антиферромагнетики. Ферромагнитными и антиферромагнитными свойствами обладают не дискретные атомы, а вещества. Если отдельные атомы переходных элементов, образующие вещество, содержат неспаренные электроны, то есть обладают парамагнетизмом, то, находясь на близком расстоянии, они взаимодействуют друг с другом, образуя домены. Если магнитные моменты доменов сонаправлены, вещество становится ферромагнетиком (Рис. 1.12 а) Ферромагентизмом обладают железо, кобальт, никель, а также редкоземельные элементы – от европия до тулия. Для тяжелых d-элементов ферромагнитные свойства не характерны, так как 4d-, 5d-орбитали сильнее взаимодействуют друг с другом, образуя ковалентные связи. К числу ферромагнетиков принадлежат низшие оксиды 3d-переходных элементов. Наиболее сильные постоянные магниты были сделаны из сплава Nd2Fe4B (G. Boebinger, A. Passner, J. Bevk, Sci. Am., 1995, v. 274, p. 59). При нагревании ферромагнетика за счет усиления тепловых колебаний атомов при определенной температуре доменная структура нарушается, и феромагнетик становится парамагнетиком. Эта температура называется температурой Кюри. Для железа она составляет 79°C, для никеля – 358°C.
Антиферромагнитные свойства объясняются противоположной ориентацией доменов (Рис. 1.12. б). Она становится возможной при взаимодействии атомов металла, содержащих неспаренные электроны, не напрямую, а опосредованно – через диамагнитную частицу (Рис. 1.12 в), например, ион кислорода или группу CN-. Антиферромагнитными свойствами обладают некоторые оксиды металлов, соли, комплексные соединения. Примером может служить комплекс [Cr2(NH2)3(NH3)6]I3 (Рис. 1.12 г), состоящий из диядерных молекул, соединенных амидными мостиками. Атомы хрома расположены на расстоянии 0,265 нм, что не позволяет их d-орбиталям перекрываться непосредственно (иначе соединение было бы диамагнитно), но делает возможным их опосредованное взаимодействие через заполненные орбитали лиганда. С ростом температуры в точке Нееля антиферромагнетик превращается в парамагнетик. Для описанного комплекса точка Нееля равна 23 °C (H. Lueken, H. Schilder, H. Jacobs, V. Zachwieja, Z. Anorg. Allg. Chem., 1995, 621, 959).
Простые вещества. d-Орбитали принимают активное участие и в формировании простых веществ. Металлическая связь в переходных металлах включает также зону перекрывания d-орбиталей различных атомов. Вклад d-орбиталей в образование связи коррелирует с теплотами атомизации металлов и температурами их плавления. Наиболее эффективно перекрывание частично заполненных орбиталей, поэтому наиболее тугоплавкие металлы расположены в середине ряда (Рис. 1.13. Перекрывание d-орбиталей с образованием энергетической зоны (а); Изменение теплот атомизации (б) и температур плавления (в) 3d-элементов). Наполовину заселенный d-подуровень обладает повышенной устойчивостью, с этим связано понижение теплоты атомизации и температуры плавления марганца по сравнению с соседними по ряду элементами. Большая величина перекрывания между 4d- и 5d-орбиталями по сравнению с 3d- приводит к возрастанию теплот атомизации и температур плавления при движении вниз по группе. Поэтому наиболее тугоплавкие металлы расположены в середине второго и третьего переходных рядов. Такими же свойствами отличаются и их бинарные соединения – бориды, карбиды.
Восстановительная активность металлов уменьшается с возрастанием порядкового номера, как по ряду, так и по группе, что объясняется ростом энергии ионизации. Таким образом, наиболее активные металлы расположены в начале 3d-ряда, а наименее активные – в конце 5d-ряда. Примером может служить взаимодействие d-металлов с кислотами. Из 3d-металлов положительный потенциал E0(Mn+/M0) имеет лишь медь, в то время как практически все элементы второго и третьего переходных рядов, за исключением кадмия, оказываются инертными по отношению к разбавленным кислотам и щелочам, растворяясь лишь в присутствии окислителя (HNO3, NOCl, KO2, KOH + KClO3) или в смеси окислителя и комплексообразователя (HNO3 + HF, O2 + KCN).
Сплавы. Твердые растворы и интерметаллиды.
Сплавы представляют собой твердые растворы на основе металлов, содержащие два, а зачатую и большее число компонентов, преимущественно металлов или неметаллов (углерод, кремний). Атомы металлов споосбны реагировать мжду собой, образуя химические соединения – интерметаллиды. Они также входят в состав многих сплавов.
Твердые растворы подразделяются на три основных типа: замещения, внедрения и вычитания. В твердых растворах замещения атомы легирующей добавки замещают атомы основного вещества-матрицы, образуя общую кристаллическую решетку. Вводимые добавки изменяют свойства сплавов (прочность, вязкость, пластичность, износостойкость). Это широко используется при создании конструкционных материалов. Образование твердых растворов сопровождается выигрышем в конфигурационной энтропии и поэтому термодинамически благоприятно. Исключения могут представлдять случаи очень сильных различий в размерах атомов компонентов – при этом введение в матрицу посторонних атомов потребует сильных ее искажений. Пороговое значение разницы в атомных радиусах компонентов твердого раствора, оенивается в 15 %. Образование непрерывных твердых растворов между компонентами сплава возможно, если металлы имеют одинаковое кристаллическое строение с близкими параметрами кристаллической решетки и одинаковый тип химической связи(Mo-W, Ag-Au), а интерметаллиды помимо этого и одинаковый стехиометрический состав (FeCr-FeV, AuCu3-PtCu3). В твердых растворах внедрения атомы растворенного вещества встраиваются в междоузлия кристаллической регетки матрицы. В этой роли чаще выступают атомы неметаллы (O, C, B, N, H, S), которые внедряются в тетраэдрические и октаэдрические пустоты кристаллических решеток металлов. Особенно широко это наблюдается в системах с переходными металлами. Твердые растворы вычитания образуются при отсутствии (дефиците) части атомов одного и, иногда, обоих компонентов. Такими примерами могут служить вюстит Fe1-xO с дефицитом атомов железа и монооксид титана, у которого в катионной и анионной подрешетках отсутствуют до 15 % атомов Ti и O. Твердые растворы вычитания наблюдаются в упорядоченных растворах нестехиометрического состава. Напрмер, избыточные атомы алюминия в твердом растворе на основе соединения CoAl (структурный тип CsCl) занимают позиции в подрешетке алюминия, оставляя вакантные позиции в подрешетке кобальта, приводя к составу Со1-хAl.
В настоящее время известно более двадцати тысяч интерметаллидов постоянного и переменного составов. Они образуются, если атомы различных компонентов сплава взаимодействуют между собой сильнее, чем атомы одного сорта. Межатомные взаимодействия особенно сильно проявляются при понижении температуры. При этом часто происходит распад высокотемпературных твердых растворов из-за их термодинамической неустойчивости с выделением новых фаз. В иинтерметаллидах наиболее часто встречаются два типа химических связей – металлическая и ковалентная. Зачастую между ними сложно провести четкую границу. Для определения характера связей изучают распределение электронной плотности между партнерами (рентгеновская дифракция), локализованные или нелокализованное распределение электронов в кристаллах (рентгеноспектральный метод), физические и механические свойства (магнетизм, электропроводность, спайность, прочность и др.), термодинамические характеристики (теплоты образования, энергии деформации и др.). Один из важных признаков стабильности интерметаллидов заключается в максимальном упорядочении расположения атомов. В формировании интерметаллидов, образованных атомами близких по размерам и электронному строению (фаз Юм-Розери), главную роль играет соотношение между числом валентных электронов и числом атомов в решетке (так называемая электронная концентрация). В случае соединений двух металлов с сильно различающимися радиусами важнейшую роль начинает играть принцип максимального заполнения атомами пространства. Примером этого служат фазы Лавеса.
ДОПОЛНЕНИЕ. Фазы Юм-Розери.
Соединения с металлической связью, называемые электронными фазами, или фазами Юм-Розери отличаются простыми и более совершенными типами кристаллической решетки с более высокими координационными числами (8, 12, 16) по сравнению с исходными металлами. Часто они образуются между металлами, атомные радиусы которых отличаются не более, чем на 15 %. Их формулы, например, AlCo, Ag3Al, LaNi5, не соответствуют обычным представлениям о валентности. Устойчивость фаз Юм-Розери определяется электронной концентрацией, то есть отношением числа валентных электронов к числу атомов в решетке. Например, рассмотрим систему Cu-Zn. Каждый атом меди имеет один валентный электрон, а у атома цинка их два. При растворении цинка в меди электронная концентрация повышается, и до значения 1,4 устойчив первичный твердый раствор – так называемая фаза α-латуни. В интервале электронных концентраций 1,4 - 1,5 устойчива β-фаза CuZn, а в интервале 1,5 -1,62, γ-латунь Cu5Zn8 со сложной кубической структурой, содержащей 52 атома в элементарной ячейке (Рис 1.14. Строение γ-латуни Cu5Zn8 – пример фазы Юм-Розери: (а) общий вид кристаллической решетки, (б) проекция на плоскость) В этих структурных типах кристаллизуются фазы в многочисленных двойных системах на основе Ag, Cu, Au, Fe, Co, Ni, Pt и др.
Cоставы указанных фаз Юм-Розери нередко не находятся в области существования равновесной фазы на диаграмме состояния системы, и их свойства не характеризуются какими-либо аномалиями. Таким образом, смысл критических значений электронной концентрации заключается в определении области , вблизи которой устойчива фаза соответствующего структурного типа.
КОНЕЦ ДОПОЛНЕНИЯ.
ДОПОЛНЕНИЕ. Фазы Лавеса
Интерметаллиды состава АВ2, в которых атомы разных металлов образуют плотную упаковку, получили название фаз Лавеса. Сейчас их известно более 300, почти во всех одним из компонентов выступает переходный металл. Для обеспечения плотной упаковки необходимо достаточно большое различие в атмоных радиусов двух металлов. Для этих фаз выведен геометрический критерий устойчивости RA/RB = √3/2 ≈ 1,225. В реальных соединениях отношения размеров колеблются от 1,05 до 1,68.
Особенность строения фаз Лавеса заключается в группировании меньших по размеру атомов (Cu в MgCu2, Ni в CuNi2, Zn в MgZn2) в сочлененные друг с другом тетраэдры Cu4, Ni4 и Zn4. Отличие структурных типов фаз Лавеса друг от друга состоит в способах сочленения этих тетраэдров. При этом создается весьма плотная упаковка: атомы Mg имеют координациионное число 16, превышающее максимальную плотность упаковки одинаковых шаров (КЧ 12). Координациионные числа меньших по размеру атомов в фазах Лавеса равно 12. Большинство фаз Лавеса не имеют широких областей твердых растворов. Устойчивость фаз Лавеса с участием переходных металлов возрастает при передаче электрона от второго компонента в частично заполненную оболочку d-металла. Такой электронный переход вносит ионный вклад в металлическую связь и сопровождается увеличением теплоты образования соединений. Фактор электронной плотности на образование фаз Лавеса сильного воздействия не имеет.
Рис. 1.15. Структура фазы Лавеса MgZn2.
КОНЕЦ ДОПОЛНЕНИЯ.
Во многих бинарных системах непереходных и переходных металлов образуются интерметаллиды эквиатомного состава АВ со структурой типа CsCl. Их образование характерно для электроположительных металлов 2=6 групп с более электроотрицательными элементами, расположенными правее 6 Группы: TiFe, BeCo, ScNi, BaZn, VTc, TiRu, MgRh. Известно более 200 таких соединений. К ним также относятся никелид титана TiNi и кадмид золота AuCd – материалы с переходами мартенситного типа, обладающие свойствами памяти формы.
Каждому составу и типу структур интерметаллидов присущи индивидуальные физические, механические, химические, термические электрические, магнитные и другие свойства. Поэтому такие материалы очень важны и интересны в практическом и научном планах и интенсивно исследуются.
Кислородные соединения. Нестехиометрия. Низшие оксиды многих переходных элементов по свойствам (электропроводность, металлический блеск) напоминают металлы, что объясняется перекрыванием частично заполненных d-орбиталей. Широко известен пример магнитного железняка Fe3O4, большие запасы которого, расположенные вблизи поверхности Земли недалеко от Курска, создают магнитное поле, нарушающее работу навигационных систем самолетов. Это соединение, как и металлическое железо, ферромагнитно и обладает заметной проводимостью. Механизм проводимости включает эстафетную передачу электрона от иона Fe2+ к Fe3+ за счет перекрывания d-орбиталей.
Монооксиды 3d-переходных металлов от титана до никеля в степени окисления +2 имеют структуру NaCl (рис. 1.16. (а) Структура NaCl, (б) Структура рутила). Многие из них представляют собой нестехиметрические соединения. Например, приготовленный обычным способом оксид железа(II) имеет состав Fe0,95O, то есть содержит недостаток атомов железа по сравнению со стехиометрией МО. Недостаток положительно заряженных ионов компенсируется окислением части атомов железа(II) в структуре до Fe3+, таким образом, при наличии вакансий в катионной подрешетке сохраняется электронейтральность. Типы дефектов, возникающих в кристаллических структурах, описаны в томе 1. Схематически они изображены на рис. (Рис.1.17. Типы дефектов).
Большинство диоксидов переходных металлов кристаллизуется в тетрагональной структуре рутила TiO2 (Рис. 1.16. Б), в кристаллической решетке которого атом металла находится в тетрагонально искаженном октаэдре из атомов кислорода.
Для переходных металлов известно лишь небольшое число триоксидов. Структура оксида ReO3 состоит из связанных общими вершинами октаэдров [ReO6] (Рис. 1.18 Структура ReO3 в виде атомов (а) и в форме полиэдров (б)). Атомы рения образуют кубическую примитивную ячейку. Близкую структуру имеет триоксид вольфрама. Если нагреть порошок триоксида вольфрама в парах натрия, то часть атомов натрия войдет в кристаллическую структуру, разместившись в центрах некоторых примитивных ячеек, образованных атомами вольфрама. Произойдет перераспределение зарядов – часть атомов вольфрама восстановится до +5, а натрий окислится до +1. За счет частичного восстановления вольфрама полученная оксидная фаза (вольфрамовая бронза) NaxWO3, (x = 0,3 – 0,9) приобретает характерные цвета (синий, красный, золотистый), металлический блеск и проводимость. Сходные по свойствам оксидные фазы известны и для других элементов (молибденовые, ванадиевые, ниобиевые, титановые бронзы).
К важным структурным типам кислородных соединений переходных элементов относятся перовскит и шпинель. Структурный тип перовскита ABХ3,(Х – О, F, S) названный так по природному минералу CaTiO3, открытому русским минералогом Л. А. Перовским, представляет собой трехслойную плотнейшую шаровую упаковку (ПШУ) из атомов Х и А, в октаэдрических пустотах которой расположены атомы В (Рис. 1.19. Структура перовскита). В структуре перовскита кристаллизуются различные соединения, в которых суммарный заряд А и В равен +6, и атомы А и В существенно различаются по размеру. Сейчас известно несколько сотен соединений со структурой перовскита.
Предполагая, что в структуре идеального перовскита атомы соприкасаются, параметр элементарной ячейки a можно выразить двумя способами (Рис. 1.20. Соотношение ионных радиусов в структуре перовскита):
 ;
;  ,
,
где r – ионные радиусы по Шеннону-Прюитту.
В реальной структуре из-за слишком малых или слишком больших размеров ионов плотность упаковки нарушается, и величины a, рассчитанные по этим уравнениям, несколько различаются. Поправка, равная отношению этих значений, носит название фактора толерантности t:
 .
.
В идеальном перовските t = 1, в титанате стронция SrTiO3 (r(Sr2+) = 0,158 нм, r(Ti4+) = 0,745 нм, r(O2-) = 0,126 нм) t = 1,002. Зная величину фактора толерантности, можно предсказать, будет ли соединение состава АВО3 иметь структуру перовскита – она возможна при 0,85 < t < 1,06.
При высокой температуре атомы В способны смещаться из центра элементарной ячейки, вызывая разделение электрических зарядов и возникновение диполя. Такие соединения называют сегнетоэлектриками. Ряд веществ со структурой перовскита, например, титанат бария BaTiO3, обладает пьезоэлектрическим эффектом. Он заключается в том, что смещение атома B, происходящее в результате механического воздействия, вызывает появление электрического поля. Пьезоэлектрики находят широкое применение в качестве преобразователей механической энергии в электрическую, например, в микрофонах, зажигалках.
Шпинелями называют сложные оксиды состава AB2O4, в которых атомы металлов занимают пустоты в трехслойной плотнейшей шаровой упаковке, образованной атомами кислорода. Этот структурный тип получил название от минерала благородная шпинель MgAl2O4. Атомы А в шпинелях представляют собой двухзарядные катионы с радиусом 0,08 – 0,11 нм, например, Mg2+, Cr2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+, Cd2+, Sn2+. Обычно они занимают одну восьмую часть всех тетраэдрических пустот в анионной подрешетке. Октаэдрические пустоты в структуре шпинели наполовину заселены атомами В – трехзарядными катионами с радиусом 0,075 – 0,09 нм (Ti3+, V3+, Cr3+, Mn3+, Fe3+, Co3+, Ni3+, Rh3+, Al3+, Ga3+, In3+). Более наглядно структуру шпинели можно представить в виде кубической объемно-центрированной ячейки из атомов А, внутри которой находятся кубы В4О4 и тетраэдры АО4 (Рис. 1.21. Структура шпинели). В некоторых шпинелях наблюдается инверсия – половина атомов В размещается в тетраэдрических пустотах, а атомы А вместе с другой половиной атомов В – в октаэдрических. Такие шпинели называют обращенными В(А,В)О4. К их числу принадлежит магнитный железняк FeIII(FeII, FeIII)O4.
Химия водных растворов. В водных растворах ионы переходных металлов присутствуют в виде сложной смеси катионных форм, в состав которой зачастую входят и полиоксогидроксокатионы, сходные с описанными во втором томе учебника на примере алюминия, свинца и висмута. Процессы, протекающие при гидролизе акваиона металла, можно условно разделить на два типа – собственно гидролиз, то есть депротонирование части молекул воды в координационной сфере металла (превращение их в гидроксо-группы), полимеризацию, приводящую к образованию гидроксильных мостиков, и поликонденсацию, связанную с образованием оксо-групп (мостиковых или терминальных) за счет отщепления молекул воды:

Конечным продуктом гидролиза является гидроксид или оксогидроксид. Степень гидролиза возрастает с увеличением заряда катиона и уменьшением его радиуса, что легко заметить, сравнивая рН осаждения различных гидроксидов. Так, например, Cd(OH)2 количественно осаждается при рН = 11, Mn(OH)2 при 8 – 9, Fe(OH)2 при 7, Cu(OH)2 и Zn(OH)2 при 6, Fe(OH)3 при 3. Трехзарядные аквакомплексы 3d-металлов [M(H2O)6]3+ существуют в водных растворах лишь в сильнокислой среде. В растворах солей трехвалентного железа, приготовленных растворением в воде без предварительного подкисления, присутствует сложная смесь гидратированных катионов: [Fe(H2O)6]3+, [Fe(H2O)5(OH)]2+, [Fe(H2O)4(OH)2]+, [Fe2(H2O)8(OH)2]4+, [Fe3(OH)5(H2O)9]4+, [Fe3O(H2O)8(OH)4]3+ и др., а также коллоидный раствор гидроксида железа. Более устойчивыми к гидролизу оказываются катионы лантанидов, имеющие гораздо больший размер. Из четырехзарядных катионов в растворе существует лишь ион тория [Th(H2O)9]4+. Практически все гидроксиды d-элементов в той или иной степени амфотерны, то есть растворяются в избытке щелочи с образованием гидроксокомплексов. Гидроксиды двух- и трехвалентных металлов 3d-ряда в крепких щелочных растворах образуют тетрагидроксокомплексы, в которых кооординационная сфера металла дополнена до шести двумя молекулами воды ([Fe(OH)4(H2O)2]–), а в сильно щелочных растворах – гексагидроксокомплексы ([Fe(OH)6]3–). У гидроксидов металлов в степени окисления +4 и выше преобладают кислотные свойства, и в щелочных растворах они существуют в форме тетраэдрических оксо-ионов, например VO43–, MnO4–. Двух- и трехзарядные оксоионы при постепенном подкислении начинают конденсироваться:
CrO42– + 2H+ = Cr2O72– + H2O,
при этом у ряда металлов (V, Nb, Тa, Mo, W) происходит изменение координационного числа и образование полиоксометаллатов (V10O286–, Mo7O246–и др).
С возможностью атомов переходных металлов легко изменять координационные числа связано образование целого класса анионных полиоксометаллатов, также называемых изополисоединениями (изополисоединения существуют и у непереходных элементов).
Дополнение. Изополисоединения.
Процессы гидролиза анионов ванадия, ниобия, тантала, молибдена и вольфрама в высших степенях окисления приводят к образованию конденсированных полиоксометаллатов, то есть многоядерных анионов, в которых практически отсутствуют гидроксо-группы, а преобладают связи M-O-M или M=O, например, [Mo7O24]6–, [Ta6O19]8– (Рис. 1.22. Строение аниона гексатанталата [Ta6O19]8– (а) представление в виде атомов, (б) представление в форме полиэдров). Чтобы подчеркнуть отсутствие в такого рода частицах каких-либо иных атомов, кроме кислорода и металла, их называют изополианионами, а кислоты и соли, в состав которых они входят – изополисоединениями. Изополианионы имеют правильную геометрическую форму и состоят из октаэдров МО6 и тетраэдров МО4, соединенных общими вершинами и ребрами, а иногда – даже гранями. Замечено, что ни в одном изополианионе не содержится октаэдров МО6 с более чем двумя концевыми атомами кислорода, то есть не более двух связей М=О (правило Липскомба). Эти короткие связи, стабилизированные pπ-dπ связыванием, то есть взаимодействием заполненных 2р-орбиталей кислорода с вакантными d-орбиталями металла, всегда направлены к внешней части полианиона, образуя слой концевых атомов кислорода, которые сильно смещены от центра октаэдра за счет pπ-dπ взаимодействия (Рис. 1.23 Образование связи М=О в изополианионе: (а) pπ-dπ взаимодействие, (б) смещение атома металла из центра октаэдра). Эти концевые атомы кислорода, благодаря pπ-dπ связыванию, утрачивают донорные свойства, в том числе способность присоединять протоны. Иными словами, эти атомы, образующие оксосвязи М=О, препятствуют образованию гидроксокомплексов, то есть гидролизу изополианиона.
В противоположность этому, у атомов р-элементов отсутствует возможность pπ-dπ свзявания, поэтому их оксосоединения оказываются неустойчивыми к гидролизу, а в водных растворах преобладают гидроксокомплексы, например, [Bi6(OH)12(H2O)12]6+, [Al13O4(OH)24(H2O)12]7+, [Ge8(OH)35]3–.
Для эффективного pπ-dπ связывания требуется, чтобы атом металла имел вакантные d-орбитали, поэтому не удивительно, что подавляющее большинство изополисоединений образовано переходными металлами в высшей степени окисления. Единственное исключение предсталяет ванадий, для которого описаны изополиоксоанионы и в степени окисления +4. Чтобы обладать способностью образовывать изополисоединения, атом должен иметь радиус, позволяющий ему иметь в водных растворах координационные числа как 4, так и 6.
Конкретные типы изополианионов описаны в главах, посвященных химии элементов. (Ссылки: М. С. Поп, Гетерополи- и изополиоксометаллаты, Новосибирск, Наука. 1990; Н.А. Добрынина, Журн. неорган. химии, 2002, 47(4), 577.)
КОНЕЦ ДОПОЛНЕНИЯ.
Дополнение: Жесткие и мягкие кислоты и основания.
В 1968 г. Р. Пирсон предложил удобную классификацию катионов и лигандов (R. G. Pearson, J. Chem. Educ., 1968, 45, 643). Мягкие кислоты и основания – это молекулы или ионы, образующие с другими частицами преимущественно ковалентные связи, например, Hg2+ – мягкая кислота, а I– – мягкое основание. Жесткие кислоты (Ca2+) и основания (F–) образуют преимущественно ионные связи. Главный постулат теории Пирсона прост: мягкие кислоты тяготеют к мягким основаниям, а жесткие – к жестким. Так, например, для кальция наиболее устойчив (и наименее растворим) фторид, а для ртути – иодид. Катионы металлов, относящиеся к жестким кислотам, образуют комплексы в основном с О-донорными лигандами, в то время как мягкие кислоты – с P-, S-донорными (Таблица 1.2).
Таблица 1.2 Мягкие и жесткие кислоты и основания
| Классы соединений | Жесткие | Промежуточные | Мягкие |
| Кислоты | Na+, K+ Ca2+, Mn2+, Al3+, Sc3+, Fe3+, Ti4+, Ln3+ | Fe2+, Co2+, Ni2+, Cu2+ | Hg2+, Hg22+, Cu+, Ag+, Au+, Pd2+, Pt2+, Rh+, Ir+ |
| Основания | F–, R2O, H2O, ROH, RO–, RCO2–, SO42–, NH3, NR3, Cl– | Br–, py | R2S, R3P, CN–, SCN–, CO, I–, R2S, S2– |
N-донорные лиганды занимают промежуточное положение. Жесткость оснований изменяется в том же порядке, что и электроотрицательность:
P3– < S2– < N3– < O2– < F–,
I– < Br– < Cl– < F–.
Исходя из постулата Пирсона, можно предсказать, что никель(II) будет образовывать более устойчивые аммиакаты, чем железо(III), а комплексы лантанидов с дикетонами будут прочнее комплексов с аминами. Устойчивость галогенидных комплексов «жестких» металлов в водных растворах возрастает от от иода к фтору, а для комплексов, образованных «мягкими» металлами – в противоположном направлении.
В конце 1980-х гг Пирсон развил свою теорию, введя понятие об абсолютной жесткости и мягкости (R. G. Pearson, Inorg. Chem., 1988, 27, 734; Inorg. Chim. Acta, 1995, 240, 93). Он определил абсолютную жесткость η как полуразность энергии ионизации I и сродства к электрону A:
 .
.
Однако количественные характеристики жесткости и мягкости (1/η) пока не получили широко применения. Другим исследователем на основании анализа констант устойчивости комплексов была составлена количественная шкала жесткости лигандов, согласно которой жесткость возрастает в ряду S2O32– < CN– » I– < Br– » SCN– < NH3 < py < OH– » CO32– < HCO3–< CH3COO– < H2O < F– (R.B. Martin, Inorg. Chim. Acta, 2002, 339, 27).
КОНЕЦ ДОПОЛНЕНИЯ
2. Комплексные соединения переходных элементов.
Важное место в химии переходных элементов занимают комплексные соединения, то есть соединения, в которых атом металла (его называют комплексообразователем) координирован одним или несколькими лигандами – молекулами или ионами, способными к самостоятельному существованию. Первоначальные сведения о комплексных соединениях приведены в томе 1. Наиболее распространенные лиганды представлены на Схеме 1:
Схема 1. Наиболее распространенные лиганды

Лиганды различаются по природе донорных атомов (чаще всего, это O, N, Cl, P) и по их числу. Число донорных атомов лиганда, химически связанных с атомом металла, называют дентатностью. Так, например, аммиак является монодентатным лигандом, этилендиамин – бидентатным, а этилендиаминтетраацетат (том 2, с. 66) – гексадентатным. Многие лиганды, содержащие более одного донорного атома, координируют один и тот же атом металла, образуя с ним хелатный цикл. Такие лиганды называют хелатирующими, а комплексы с ними – хелатами. Хелаты оказываются термодинамически и кинетически более устойчивыми, чем аналогичные комплексы с монодентатными лигандами. Это явление называют хелатным эффектом. В качестве примера приведем значения констант устойчивости двух комплексов – с аммиаком и этилендиамином:
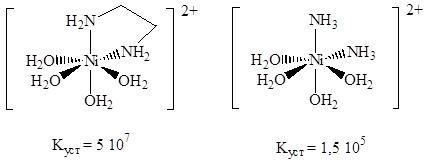
Увеличение константы устойчивости комплекса с этилендиамином объясняется возрастанием энтропии при увеличении числа частиц в растворе в результате реакции замещения:
[Ni(H2O)6]2+ + en = [Ni(H2O)4en]2+ + 2H2O
При образовании аммиаката число частиц в растворе не изменяется:
[Ni(H2O)6]2+ + 2NH3 = [Ni(H2O)4(NH3)2]2+ + 2H2O.
Другой причиной хелатного эффекта служит крайне низкая скорость раскрытия хелатного цикла (k1 << k–1). Диссоциация хелатирующего лиганда протекает в несколько стадий, на первой из которых лиганд металл еще координирован одним из донорных атомов лиганда. Это не позволяет другому донорному атому отойти от металла на значительное расстояние и делает более вероятным повторное образование хелатного цикла:
 Некоторые простые анионные лиганды, например, OH–, F–, Cl–, O2–, CO часто выступают в роли мостиковых, связывая сразу несколько металлов при помощи одного донорного атома. В специальной литературе это обозначают введением в название комплекса перед названием лиганда греческой буквы μ индексом, указывающим количество связанных атомов металла. Лиганды, содержащие несколько донорных центров, способны образовывать мостики и другого рода, обозначаемые буквой η:
Некоторые простые анионные лиганды, например, OH–, F–, Cl–, O2–, CO часто выступают в роли мостиковых, связывая сразу несколько металлов при помощи одного донорного атома. В специальной литературе это обозначают введением в название комплекса перед названием лиганда греческой буквы μ индексом, указывающим количество связанных атомов металла. Лиганды, содержащие несколько донорных центров, способны образовывать мостики и другого рода, обозначаемые буквой η:

ДОПОЛНЕНИЕ Комплексы с макроциклическими лигандами
Макроциклическими называют циклические полидентатные лиганды. К их числу относят циклические полиамины, краун-эфиры, криптанды (см. том 2, глава 2) и порфирины. В зависимости от размера полости в лиганде и от ионного радиуса металла образуются комплексы, в которых центральный атом находится внутри макроцикла или над ним (Рис.1.24. Строение комплексов щелочных металлов с краун-эфирами: (а) [Li(12-краун-4)Cl], (б) [Na(бензо-15-краун-5)(H2O)]I, (в) [K(дибензо-18-краун-6)I], (г) Na[Cs(18-краун-6)2]). Например, иону Li+ оптимально соответствует размер полости в 12-краун-4, Na+ - в 15-краун-5, К+ - в 18-краун-6. В этих комплексных ионах атомы металла лишь незначительно выступают из плоскости макроцикла, что приводит к высоким значениям констант устойчивости. В то же время полость в 18-краун-6 не способна вместить ионы тяжелых щелочных металлов, например, цезия. Поэтому цезиевый комплекс менее устойчив. Константы устойчивости комплексов с макроциклическими лигандами выше аналогичных значений для нециклических лигандов с равным числом донорных атомов и сходной структурой. Это явление получило название макроциклического эффекта. В случае, если радиус атома металла идеально соответствует размеру полости, образование комплекса с макроциклом энергетически более благоприятно, даже несмотря на уменьшение энтропии:

Если размер полости не позволяет атому металла войти в нее, главное значение приобретает энтропийный фактор. Нециклический лиганд находится в постоянном вращении вокруг одинарных связей, для образования комплекса он должен приобрети определенную конформацию, то есть уменьшить энтропию. В случае макроциклического лиганда такая конформация уже зафиксирована заранее, т. к. вращение в цикле невозможно.(E.C. Constable, Coordination Chemistry of Macrocyclic Compounds, Oxford University Press, Oxford, 1999)
КОНЕЦ ДОПОЛНЕНИЯ
Теория кристаллического поля (ТКП).
Простым и наглядным способом описания строения комплексных соединений служит теория кристаллического поля (ТКП). Она исходит из предположения об электростатическом взаимодействии d-орбиталей атома металла с лигандами, рассматриваемыми как точечные отрицательные заряды. В поле сферической симметрии все пять d-орбиталей вырождены (обладают одинаковыми энергиями). В октаэдрическом поле из-за из-за понижения симметрии вырождение снимается, и орбитали разделяются на две группы – обладающие большей (dz2, dx2-y2, их обозначают eg) и меньшей (dxy, dxz, dyz, их обозначают t2g) энергией. Разница в энергиях орбиталей представляет собой параметр расщепления октаэдрическим полем, обозначаемый Δo, или 10Dq, как это принято в спектроскопии. f-Орбитали в октаэдрическом поле расщепляются на три энергетических уровня, обозначаемые a2g, t1g, t2g (Рис. 1.25. (а) Форма f-орбиталей (слева xyz, z(x2-y2), y(z2-x2) и x(z2-x2), справа x3, y3, z3), (б) Расщепление f-орбиталей в октаэдрическом поле). p-Орбитали не расщепляются.
Выигрыш энергии при переходе из сферического поля в октаэдрическое, выраженный в единицах Δo, называют энергией стабилизации кристаллическим полем (ЭСКП). Заполнение t2g- и eg-орбиталей электронами d1-d3 и d8-d10 происходит в полном соответствии с правилом Хунда. При электронных конфигурациях d4 – d7 возможна альтернатива – электроны могут спариваться, оставаясь на t2g-уровне с более низкой энергией (это приводит к образованию низкоспинового комплекса) или заполняя энергетически менее устойчивый eg-уровень (такой комплекс называется высокоспиновым) (Рис. 1.26. Схема образования высокоспинового и низкоспинового комплексов). Какой из двух процессов реализуется, зависит от соотношения величин энергии спаривания P, то есть энергии, затрачиваемой на спаривание электронов, и параметра расщепления Δo. Если Δo>P, образуется низкоспиновый комплекс, а если Δo<P – высокоспиновый. Энергия спаривания зависит от заряда иона: для трехзарядных катионов она выше, чем для двухзарядных (Табл. 1.3).
Таблица 1.3.
Значения энергий спаривания некоторых ионов в газовой фазе*
| Электронная конфигурация | Энергия спаривания для M2+ | Энергия спаривания для M3+ | ||
| M2+ | Р, см–1 ** | M3+ | Р, см–1 ** | |
| d4 d5 d6 d7 | M2+ M2+ M2+ M2+ | M3+ M3+ M3+ M3+ |
____
* В комплексных соединениях энергия спаривания на 15 – 30 % ниже из-за ковалентного вклада в связь металл-лиганд.
** 1 кДж = 83 см–1
Величина Δo резко возрастает при переходе от 3d-элементов к 4d и 5d, т.к. с ростом размеров d-орбиталей усиливается их электростатическое взаимодействие (отталкивание) с лигандами. Все комплексы тяжелых (4d, 5d) переходных металлов, независимо от природы лиганда, низкоспиновые. Энергия расщепления растет с увеличением степени окисления центрального атома. Чем выше степень окисления металла, тем выше заряд катиона, тем меньше его радиус и сильнее отталкивание между лигандами и eg-электронами (Табл. 1.4).
Таблица1.4.
Энергии расщепления и окраска аквакомплексов d-элементов
| Электронная конфигурация | Аквакомплексы 3d-элементов | Аквакомплексы 4d-, 5d-элементов | |||
| Mn+ | Δo, см–1 | Окраска | Мn+ | Δo, см–1 | Окраска |
| d1
d2
d3
d4
d5
d6
d7
d8
12 Дата добавления: 2016-01-03; просмотров: 7931; |
Генерация страницы за: 0.083 сек.