Метаболизм галактозы
Галактоза образуется в кишечнике в результате гидролиза лактозы.
Нарушение метаболизма галактозы проявляется при наследственном заболевании – галактоземии. Оно является следствием врожденного дефекта фермента гексозо-1-фосфат-уридилилтрансферазы. Галактоземия проявляется вскоре после рождения, как только ребенок начинает получать молоко, в виде рвоты, диареи, дегидратации, уменьшении массы тела, желтухи. В крови, моче и тканях повышается концентрация галактозы и галактозо-1-фосфата. Вскоре после рождения развивается катаракта хрусталика, гепатомегалия, поражение почек и головного мозга, в тяжелых случаях возможен летальный исход.
В гораздо более редких случаях причиной развития галактоземии могут быть наследственные дефекты других ферментов метаболизма галактозы – галактокиназы и УДФ-глюкозо-4-эпимеразы. Клинические проявления этих дефектов менее выражены.
Метаболизм лактозы
Лактоза, дисахарид содержится только в молоке и состоит из галактозы и глюкозы. Лактоза синтезируется только секреторными клетками желез млекопитающих в период лактации. Она присутствует в молоке в количестве от 2 % до 6 % в зависимости от вида млекопитающих.
Синтез лактозы идет на основе глюкозы и УДФ-галактозы. Благодаря обратимому действию фермента УДФ-глюкозо-4-эпимеразы имеет место взаимопревращение:
 УДФ-глюкоза УДФ-галактоза.
УДФ-глюкоза УДФ-галактоза.
Далее фермент лактозосинтетаза осуществляет реакцию конденсации:
лактозо-
синтетаза
 УДФ-галактоза+глюкоза лактоза + УДФ.
УДФ-галактоза+глюкоза лактоза + УДФ.
Лактозосинтетаза состоит из двух субъединиц: каталитической и модифицирующей. Модифицирующая субъединица представляет собой α-лактальбумин.
Нарушения переваривания лактозы в кишечнике могут быть наследственными и приобретенными. Наследственный дефицит лактазы встречается относительно редко. После приема молока наблюдаются рвота, диарея, спазмы и боли в животе, метеоризм. Симптомы развиваются сразу после рождения. Вторая разновидность данной патологии – недостаточность лактазы вследствие снижения экспрессии гена фермента в онтогенезе. Характерна для взрослых и детей старшего возраста. Является следствием возрастного снижения количества лактазы. Симптомы непереносимости молока аналогичны наследственной форме дефицита лактозы. Кроме того выделяют недостаточность лактазы вторичного характера, причиной которой могут быть кишечные заболевания, операции на ЖКТ.
ГЛАВА 17
пути МЕТАБОЛИЗМА ГЛЮКОЗЫ
Глюкоза является основным метаболитом и транспортной формой углеводов в организме человека и животных. Источниками глюкозы являются углеводы пищи, гликоген тканей и процесс глюконеогенеза в печени и корковом веществе почек. Для включения глюкозы в метаболизм она должна фосфорилироваться с образованием глюкозо-6-фосфата (Г-6-Ф), который далее может превращаться по различным метаболическим путям. На Рис. 17.1. представлены основные пути метаболизма глюкозы.
Гликолиз
Гликолиз – главный путь катаболизма глюкозы путем последовательных ферментативных превращений до лактата (без потребления кислорода – анаэробный гликолиз) или через окислительное декарбоксилирование пирувата до СО2 и Н2О (в присутствии кислорода – аэробный гликолиз).
Процесс аэробного гликолиза включает несколько стадий:
1. Аэробный гликолиз – процесс окисления глюкозы с образованием двух молекул пирувата;
2. Общий путь катаболизма, включающий окислительное декарбоксилирование пирувата до ацетил КоА и его дальнейшее окисление в цикле трикарбоновых кислот;
3. Цепь тканевого дыхания, сопряженная с реакциями дегидрирования, происходящими в процессе распада глюкозы.
Суммарный выход АТФ при окислении 1 моль глюкозы до СО2 и Н2О составляет 38 моль.
Гликоген
 | |
5 6
1, 2, 3 4
 Глюкоза Г-6-Ф ПФП
Глюкоза Г-6-Ф ПФП
7
71, 2, 3
 Этанол3Пируват Лактат
Этанол3Пируват Лактат
 1 7
1 7
Ацетил-КоА
ЦТК
 |  |
СО2 Н2О
1 – аэробный гликолиз;2 – анаэробный гликолиз;3 – спиртовое брожение;
4 – пентозофосфатный путь;5 – синтез гликогена;6 – распад гликогена;
7 – глюконеогенез.
Рис. 17.-1. Общая схема путей метаболизма глюкозы.
Анаэробный гликолиз – процесс расщепления глюкозы с образованием в качестве конечного продукта лактата. Этот процесс протекает без использования кислорода и поэтому не зависит от работы митохондриательной сети. АТФ здесь образуется за счет реакций субстратного фосфорилирования. Баланс АТФ при анаэробном гликолизе составляет 2 моль в расчете на 1 моль глюкозы.
Аэробный гликолиз происходит во многих органах и тканях и служит основным, хотя и не единственным, источником энергии для жизнедеятельности.
Кроме энергетической функции гликолиз может выполнять и анаболические функции. Метаболиты гликолиза используются для синтеза новых соединений. Так, фруктозо-6-фосфат и глицеральдегид-3-фосфат участвуют в образовании рибозо-5-фосфата – структурного компонента нуклеотидов. 3-фосфоглицерат может включаться в синтез аминокислот, таких как серин, глицин, цистеин. В печени и жировой ткани ацетил-КоА, образующийся из пирувата, используется как субстрат при биосинтезе жирных кислот, холестерина.
Анаэробный гликолиз активизируется в мышцах при интенсивной мышечной работе, происходит в эритроцитах (в них отсутствуют митохондрии), а также в разных условиях ограниченного снабжения их кислородом (спазм и тромбоз сосудов, формирование атеросклеротических бляшек).
Пентозофосфатный путь (ПФП)
ПФП, называемый также гексозомонофосфатным шунтом, служит альтернативным путем окисления глюкозо-6-фосфата. По ПФП в печени метаболизируется до 33 % всей глюкозы, в жировой ткани – до 20 %, в эритроцитах – до 10 %, в мышечной ткани – менее 1 %. Наиболее активно ПФП протекает в жировой ткани, печени, коре надпочечников, эритроцитах, молочной железе в период лактации, семенниках. ПФП состоит из 2 фаз (частей) – окислительной и неокислительной.
В окислительной фазе глюкозо-6-фосфат необратимо окисляется в пентозу – рибулозо-5-фосфат, и образуется восстановленный НАДФН2. В неокислительной фазе рибулозо-5-фосфат обратимо превращается в рибозо-5-фосфат, метаболиты гликолиза и другие фосфорилированные сахара.
Биологическая роль ПФП:
1. Наработка восстановленного НАДФН2 для восстановительных биосинтезов (жирных кислот, холестерина и т. д.).
2. Синтез пентозофосфатов для образования нуклеиновых кислот и некоторых коферментов.
3. Синтез моносахаридов с числом углеродных атомов от 3 до 8.
4. Обезвреживание ксенобиотиков – необходим НАДФН2.
5. В растениях – участие в темновой фазе фотосинтеза как акцептор СО2.
ПФП не приводит к синтезу АТФ, т. е. не выполняет энергетическую функцию.
Глюконеогенез (ГНГ)
Глюконеогенез – это синтез глюкозы из неуглеводных предшественников. Основной функцией ГНГ является поддержание уровня глюкозы в крови в период длительного голодания и интенсивных физических нагрузок. Процесс протекает в основном в печени и менее интенсивно в корковом веществе почек, а также в слизистой оболочке кишечника. Эти ткани могут обеспечивать синтез 80-100 г глюкозы в сутки.
Первичными субстратами (предшественниками) в ГНГ являются лактат, глицерол, большинство аминокислот. Включение этих субстратов в ГНГ зависит от физиологического состояния организма.
Лактат – продукт анаэробного гликолиза, образуется в работающих мышцах и, непрерывно в эритроцитах. Таким образом, лактат используется в ГНГ постоянно. Глицерол высвобождается при гидролизе жиров в жировой ткани в период голодания или при длительной физической нагрузке. Аминокислоты образуются в результате распада мышечных белков и выполняются в ГНГ при длительном голодании или продолжительной мышечной работе. Аминокислоты, которые при катаболизме превращается в пируват или метаболиты цикла трикарбоновых кислот, могут рассматриваться как потенциальные предшественники глюкозы и носят название гликогенных.
Из всех аминокислот, поступающих в печень, примерно 30 % приходится на долю аланина. Это объясняется тем, что при расщеплении мышечных белков образуются аминокислоты, многие из которых превращаются сразу в пируват или сначала в оксалоацетат, а затем в пируват. Последний превращается в аланин, приобретая аминогруппу от других аминокислот. Аланин из мышц переносится кровью в печень, где снова преобразуется в пируват, который частично окисляется и частично включается в ГНГ. Такая последовательность превращений приводит к формированию глюкозо-аланинового цикла.
Мышцы Кровь Печень
 | |
 Глюкоза Глюкоза Глюкоза
Глюкоза Глюкоза Глюкоза
| | |
Гликолиз ГНГ
Пируват Пируват
| | |
 Аланин Аланин Аланин
Аланин Аланин Аланин
Рис. 17.2. Глюкозо-аланиновый цикл.
Путь глюкуроновой кислоты
Он относится к вторичным путям метаболизма глюкозы.
АТФ АДФ

 Глюкоза Глюкозо-6-фосфат
Глюкоза Глюкозо-6-фосфат
гексокиназафосфоглюкомутаза
УТФ РР

 Глюкозо-1-фосфат УДФ-глюкоза
Глюкозо-1-фосфат УДФ-глюкоза
УДФ-глюкозо-
Пирофосфорилаза
Н2О 2НАД 2НАДН●+Н+
 | |||
| | |||
 УДФ-глюкуронат
УДФ-глюкуронат
 УДФ-глюкозо-
УДФ-глюкозо-
Дегидрогеназа
Н2О
Протеогликаны
Детоксикация
УДФ
Глюкуронат
 НАДФН2глюкуронад
НАДФН2глюкуронад
редуктаза
НАДФ
 ПФП ксилулоза L-гулонат
ПФП ксилулоза L-гулонат
Альдоно-
Н2О Н2О лактоназа
 L-аскорбиновая L-гулонолактон
L-аскорбиновая L-гулонолактон
Кислота
Гулонолактон-
Оксидаза
Доля глюкозы, отвлекаемой на метаболизм по пути глюкуроновой кислоты очень невелика по сравнению с большим ее количеством, расщепляемым в процессе гликолиза или синтеза гликогена. Однако продукты этого вторичного пути жизненно необходимы организму.
УДФ-глюкуронат способствует обезвреживанию некоторых чужеродных веществ и лекарственных препаратов. Кроме того, он служит предшественником Д-глюкуронатных остатков в молекулах гиалуроновой кислоты и гепарина. В организме человека, морской свинки и некоторых видов обезьян аскорбиновая кислота (витамин С) не синтезируется, так как у них отсутствует фермент гулонолактон-оксидаза. Эти виды должны получать весь необходимый им витамин С с пищей.
ГЛАВА18
ОБМЕН ГЛИКОГЕНА
Гликоген – основной резервный полисахарид в животных тканях. Он представляет собой разветвленный гомополимер глюкозы, в котором остатки глюкозы соединены в линейных участках α-1,4-гликозидными связями, а в точках ветвления – α-1,6- гликозидными связями. Эти связи образуются примерно с каждым десятым остатком глюкозы, то есть точки ветвления в гликогене встречаются примерно через каждые десять остатков глюкозы. Так возникает древообразная структура с молекулярной массой 105 – 108 Да и выше. При полимеризации глюкозы снижается растворимость образующейся молекулы гликогена и, следовательно, её влияние на осмотическое давление в клетке. Это обстоятельство объясняет, почему в клетке депонируется гликоген, а не свободная глюкоза.
После приема пищи, богатой углеводами, запас гликогена в печени может составлять примерно 5 % от её массы. В мышцах запасается около 1 % гликогена, однако масса мышечной ткани значительно больше и поэтому общее количество гликогена в мышцах приблизительно в 2 раза больше, чем в печени. Гликоген может синтезироваться во многих клетках, например в нейронах, макрофагах, адипоцитах, но содержание его в этих тканях незначительно. В организме может содержаться до 400 г гликогена. Распад гликогена печени служит в основном для поддержания уровня глюкозы в крови в постабсорбтивном периоде. Поэтому содержание гликогена печени служит в основном для поддержания уровня глюкозы в крови в постабсорбтивном периоде. Поэтому содержание гликогена в печени изменяется в зависимости от режима питания. Гликоген мышц служит резервом глюкозы – источника энергии при мышечном сокращении. Мышечный гликоген не используется для поддержания уровня глюкозы в крови.
Синтез гликогена (гликогеногенез)
Гликоген синтезируется в период пищеварения (через 1-2 часа после приема углеводный пищи). Синтез гликогена из глюкозы, как и любой анаболический процесс, является эндергоническим, т. е. требует затрат энергии.
Синтез гликогена включает 4 этапа:
1. Фосфорилирование глюкозы до глюкозо-6-фосфата при участии гексокиназы или глюкокиназы.
2. Активация первого углеродного атома с образованием активной формы – УДФ – глюкозы.
3. Образование α-1,4-гликозидных связей. В присутствии «затравки» гликогена (молекулы, включающей не менее 4 остатков глюкозы) фермент гликогенсинтаза присоединяет остатки глюкозы из УДФ-глюкозы к С4-атому концевого остатка глюкозы в гликогене, образуя α-1,4-гликозидную связь.
4. Образование α-1,6-гликозидных связей (точки ветвления молекулы). Образование их осуществляется амилозо-1,4 → 1,6-трансглюкозидазой (ветвящий или бранчинг фермент). Когда длина линейного участка цепи включает минимально 11 остатков глюкозы, этот фермент переносит фрагмент (1 → 4) цепи с минимальным количеством 6 остатков глюкозы на соседнюю цепь или на несколько участков глюкозы дальше, образуя α-1,6-гликозидную связь. Таким образом, образуется точка ветвления. Ветви растут путем последовательного присоединения (1 – 4)-глюкозильных единиц и дальнейшего ветвления.
Гликогенсинтаза – регуляторный фермент, существующий в двух формах: 1. – дефосфорилированной, активной (форма а); 2. – фосфорилированной, неактивной (форма b). Активная форма образуется из неактивной под действием фосфатазы гликогенсинтазы при дефосфорилировании. Превращение активной формы в неактивную происходит при участии протеинкиназы путем фосфорилирования за счет АТФ.
Глюкагон
Адреналин
Å
 Аденилатциклаза Аденилатциклаза
Аденилатциклаза Аденилатциклаза
не активная активная
Å
 ц-АМФ АТФ
ц-АМФ АТФ
Å
Протеинкиназа Протеинкиназа
не активная активная
АТФ АДФ


 Гликогенn Гликогенсинтаза Гликогенсинтаза
Гликогенn Гликогенсинтаза Гликогенсинтаза

 +УДФ-глюкоза a b
+УДФ-глюкоза a b
 Гликоген(n+1)
Гликоген(n+1)
Протеинфосфатаза
Рн
Å
Инсулин
Рис. 18.-1. Регуляция активности гликогенсинтазы.
Распад гликогена может проходить двумя путями.
1. Гидролитический – при участии амилазы с образованием декстринов и даже свободной глюкозы.
2. Фосфоролитический – под действием фосфорилазы и образованием глюкозо-1-фосфата. Это основной путь распада гликогена.
Фосфорилаза – сложный регуляторный фермент, существующий в двух формах – активной и неактивной. Активная форма (фосфорилаза а) – это тетрамер, в котором каждая субъединица соединена с остатком ортофосфата через гидроксильную группу серина. Под действием фосфатазы фосфорилазы происходит дефосфорилирование, отщепление 4 молекул фосфорной кислоты, и фосфорилаза а превращается в неактивную форму – фосфорилазу b, распадаясь на две димерные молекулы. Фосфорилаза b активируется путем фосфорилирования остатков серина за счет АТФ под действием фермента киназы фосфорилазы. В свою очередь этот фермент также существует в двух формах. Активная киназа фосфорилазы – фосфорилированный фермент, превращается в неактивную форму под действием фосфатазы. Активация киназы фосфорилазы осуществляется путем фосфорилирования за счет АТФ в присутствии ионов Mg2+ протеинкиназой.
Регуляция синтеза и распада гликогена носит каскадный характер и происходит путем химической модификации ферментов.
Поскольку синтез и распад гликогена протекают по разным метаболическим путям, эти процессы могут контролироваться реципрокно. Влияние гормонов на синтез и распад гликогена осуществляется путем изменения в противоположных направлениях активности двух ключевых ферментов: гликогенсинтазы и гликогенфосфорилазы с помощью их фосфорилирования и дефосфорилирования. Инсулин стимулирует синтез гликогена и тормозит распад, адреналин и глюкагон обладают противоположным эффектом.
Нарушения обмена гликогена
Гликогеновые болезни – группа наследственных нарушений в основе которых лежит снижение или отсутствие активности ферментов, катализирующих реакции синтеза или распада гликогена. К данным нарушениям относятся гликогенозы и агликогеноз.
Гликогенозы – заболевания, обусловленные дефектом ферментов участвующих в распаде гликогена. Они проявляются или необычной структурой гликогена, или его избыточным накоплением в печени, мышцах и других органах. В настоящее время предлагается деление гликогенозов на 2 группы: печеночные и мышечные.
Печеночные формы гликогенозов проявляются в нарушении использования гликогена для поддержания уровня глюкозы в крови. Общий симптом этих форм – гипогликемия в постабсорбтивный период. К этой группе относятся гликогенозы I, III, IY, YI, IX и X типов по нумерации Кори.
Мышечные формы гликогенозов характеризуются нарушениями в энергоснабжении скелетных мышц. Эти болезни проявляются при физических нагрузках и сопровождаются болями и судорогами в мышцах, слабостью и тыстрой утомляемостью. К ним относятся гликогенозы Y и YII типов.
Агликогеноз (гликогеноз О по классификации) – заболевание, возникающее в результате дефекта гликогенсинтазы. В печени и других тканях наблюдается очень низкое содержание гликогена. Это проявляется резко выраженной гипогликемией в постабсорбтивном периоде. Характерным симптомом являются судороги, особенно по утрам. Болезнь совместима с жизнью, но больные дети нуждаются в частом кормлении.
ГЛАВА 19
ЛИПИДЫ ТКАНЕЙ, ПЕРЕВАРИВАНИЕ И ТРАНСПОРТ ЛИПИДОВ
Липиды – неоднородная в химическом отношении группа веществ биологического происхождения, общим свойством которых является гидрофобность и способность растворяться в неполярных органических растворителях. Существует несколько классификаций липидов: физико-химическая, биологическая или физиологическая и структурная. Наиболее сложной является структурная классификация, основанная на структурных особенностях этих соединений. Согласно этой классификации, все липиды делятся на омыляемые и неомыляемые. К омыляемым относят те соединения, которые при щелочном гидролизе образуют соли жирных кислот (мыла), неомыляемые же липиды щелочному гидролизу не подвергаются.
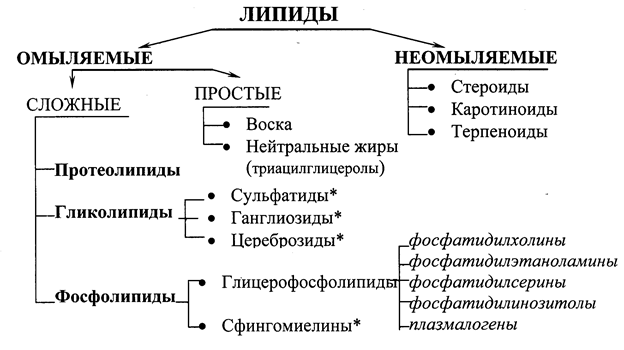
Рис. 19.1. Классификация липидов.
*В некоторых классификациях сфингомиелины, сульфатиды, ганглиозиды и цереброзиды объединяют в группу сфинголипидов, так как все они содержат аминоспирт сфингозин.
Разделение липидов по физико-химическим свойствам учитывает степень их полярности. По этому признаку липиды делятся на нейтральные или неполярные (не имеющие заряда), и полярные (несущие заряд), например, фосфолипиды и жирные кислоты. По физиологическому значению липиды делятся на резервные и структурные. Резервные липиды депонируются в больших количествах и затем расходуются для энергетических нужд организма. К резервным липидам относятся триацилглицеролы (ТАГ). Все остальные липиды можно отнести к структурным. Они не имеют особой энергетической ценности, но участвуют в построении биологических мембран и защитных покровов.
Характерным структурным компонентом большинства липидов являются жирные кислоты. Это длинноцепочечные органические кислоты, состоящие из 4-24 углеродных атомов и содержащие одну карбоксильную группу и длинный неполярный углеводородный «хвост». В составе ТАГ жирные кислоты выполняют функцию депонирования энергии. В составе фосфолипидов и сфинголипидов жирные кислоты образуют внутренний гидрофобный слой мембран, определяя его свойства. В клетках и тканях жирные кислоты встречаются в ковалентно связанной форме в составе липидов различных классов. В свободном состоянии жирные кислоты в организме содержатся в небольшом количестве, например в крови, где они транспортируются в комплексе с белком альбумином. Большинство жирных кислот образуется в организме человека, однако линолевая и линоленовая не синтезируются, поэтому обязательно должны поступать с пищей. Эти кислоты называются незаменимыми или эссенциальными. К ним относят и арахидоновую кислоту, которая может синтезироваться в организме из линолевой при достаточном поступлении последней.
Функции липидов важны и разнообразны:
- субстратно-энергетическая: жир служит в организме весьма эффективным источником энергии либо при непосредственном использовании, либо потенциально – в форме запасов жировой ткани;
- структурная (пластическая): липиды в виде комплекса с белками являются структурными элементами мембран клеток и клеточных органелл;
- транспортная: являясь одним из основных компоненнтов клеточных мембран, липиды определяют транспорт веществ в клетки;
- механическая защита: жировая прослойка предохраняет тело и органы от механических повреждений;
- теплоизолирующая: благодаря выраженной низкой термопроводимости, липиды сохраняют тепло в организме;
- электроизолирующая: липиды являются электроизолирующим материалом, участвуя таким образом в передаче нервного импульса и, соответственно, в функционировании нервной системы;
- эмульгирующая: фосфоглицеролы и желчные кислоты стабилизируют эмульсию на поверхности раздела фаз масло-вода;
- гормональная (регуляторная): стероидные гормоны, синтезируемые из холестерола, участвуют в регуляции водно-солевого обменов, половых функций; эйкозаноиды, производные полиеновых жирных кислот, вызывают разнообразные биологические эффекты;
- витаминная: в натуральных пищевых жирах содержатся жирорастворимые витамины и незаменимые жирные кислоты;
- растворяющая: одни липиды являются растворителями для других липидных веществ.
Липиды тканей человека.Липиды составляют около 10-12% массы тела человека. В среднем в теле взрослого человека содержится около 10-12 кг липидов, из них 2-3 кг приходится на структурные липиды, а остальное количество – на резервные. Основная масса резервных липидов (около 98%) сосредоточена в жировой ткани и представлена ТАГ. Эти липиды являются источником потенциальной химической энергии, доступной в периоды голодания.
Содержание липидов в тканях человека существенно различается. В жировой ткани они составляют до 75% сухого веса. В нервной ткани липидов содержится до 50% сухого веса, основные из них фосфолипиды и сфингомиелины (30%), холестерол (10%), ганглиозиды и цереброзиды (7%). В печени общее количество липидов в норме не превышает 10-14%.
Жирные кислоты, характерные для организма человека, содержат чётное число атомов углерода, чаще всего – от 16 до 20. Основной насыщенной жирной кислотой в липидах человека является пальмитиновая (до 30-35%). Ненасыщенные жирные кислоты представлены моноеновыми и полиеновыми. Двойные связи в жирных кислотах в организме человека имеют цис-конфигурацию Жиры и фосфолипиды организма при нормальной температуре тела имеют жидкую консистенцию, так как количество ненасыщенных жирных кислот преобладает над насыщенными. В фосфолипидах мембран ненасыщенных кислот может быть до 80-85%, а в составе подкожного жира – до 60%.
Липиды пищи, их переваривание и всасывание.Взрослому человеку требуется от 70 до 145 г липидов в сутки в зависимости от трудовой деятельности, пола, возраста и климатических условий. При рациональном питании жиры должны обеспечивать не более 30% от общей калорийности рациона. Жидкие жиры (масла), содержащие в своем составе незаменимые жирные кислоты, должны составлять не менее одной трети жиров пищи.
В ротовой полости и желудке взрослого человека нет ферментов и условий для переваривания липидов. Основное место расщепления липидов – тонкий кишечник. Для увеличения поверхности соприкосновения с гидрофильными ферментами жиры должны эмульгироваться (разбиться на мелкие капли). Эмульгирование происходит под действием солей желчных кислот. Эмульгированию также способствует перистальтика кишечника и выделение пузырьков СО2, происходящее при нейтрализации кислого содержимого желудка бикарбонатом, выделяющимся в составе сока поджелудочной железы.
Основная масса липидов пищи представлена ТАГ, меньше фосфолипидами (ФЛ) и стероидами. Постадийный гидролиз ТАГ осуществляется панкреатической липазой. Она секретируется в кишечник в неактивном виде и активируется колипазой и желчными кислотами. Панкреатическая липаза гидролизует жиры преимущественно в положениях 1 и 3, поэтому основными продуктами гидролиза являются глицерол, свободные жирные кислоты, моноацилглицеролы.
Фосфолипиды гидролизуются панкреатическими фосфолипазами А1, А2, С и D. Продуктами переваривания являются глицерол, жирные кислоты, фосфорная кислота и азотистые спирты (холин, этаноламин, серин, инозитол). Эфиры холестерола (ЭХЛ) расщепляются панкреатической холестеролэстеразой на холестерол (ХЛ) и жирные кислоты. Активность фермента проявляется в присутствии желчных кислот.
Всасывание липидов происходит в проксимальной части тонкого кишечника. 3-10% жиров пищи всасывается без гидролиза в виде триацилглицеролов. Основная же часть липидов всасывается лишь в виде продуктов расщепления. Всасывание гидрофильных продуктов переваривания (глицерол, жирные кислоты с числом углеродных атомов менее 12, фосфорная кислота, холин, серин, этаноламин и др.) происходит самостоятельно, а гидрофобные (ХЛ, длинноцепочечные жирные кислоты, ди- и моноглицеролы) всасываются в составе мицелл. Главную роль в образовании мицелл играют желчные кислоты. Мицелла – это сферический комплекс, в центре которого находятся транспортируемые гидрофобные продукты переваривания, окруженные желчными кислотами. Мицеллы сближаются со щеточной каймой клеток слизистой оболочки кишечника, и липидные компоненты мицелл диффундируют через мембраны внутрь клеток. Вместе с продуктами гидролиза липидов всасываются жирорастворимые витамины и соли желчных кислот. Желчные кислоты далее по воротной вене возвращаются в печень, а липидные компоненты включаются в процесс ресинтеза. В ресинтезе ТАГ участвуют не только жирные кислоты, всосавшиеся из кишечника, но и жирные кислоты, синтезированные в организме, поэтому по составу ресинтезированные жиры отличаются от полученных с пищей. Однако возможности адаптировать в процессе ресинтеза состав пищевых жиров к составу жиров организма человека ограничены, поэтому при поступлении жиров с необычными жирными кислотами в адипоцитах появляются жиры, содержащие такие кислоты. В клетках слизистой оболочки кишечника происходит синтез ФЛ, а также образование эфиров холестерола, катализируемое ацилхолестеролацилтрансферазой.
Транспорт липидов.Липиды в водной среде нерастворимы, поэтому для их транспорта в организме образуются комплексы липидов с белками – липопротеины (ЛП). Различают экзо- и эндогенный транспорт липидов. К экзогенному относят транспорт липидов, поступивших с пищей, а к эндогенному – перемещение липидов, синтезированных в организме.
Существует несколько типов ЛП, но все они имеют сходное строение – гидрофобное ядро и гидрофильный слой на поверхности. Гидрофильный слой образован белками, которые называют апопротеинами, и амфифильными молекулами липидов – фосфолипидами и холестеролом. Гидрофильные группы этих молекул обращены к водной фазе, а гидрофобные – к ядру, в котором находятся транспортируемые липиды. Апопротеины выполняют несколько функций:
· формируют структуру липопротеинов (например, В-48 – основной белок ХМ, В-100 – основной белок ЛПОНП, ЛППП, ЛПНП);
· взаимодействуют с рецепторами на поверхности клеток, определяя, какими тканями будет захватываться данный тип липопротеинов (апопротеин В-100, Е);
· являются ферментами или активаторами ферментов, действующих на липопротеины (С-II – активатор ЛП-липазы, А-I – активатор лецитин:холестеролацилтрансферазы).
Таблица 19.1.
Характеристика и состав липопротеинов
| Типы ЛП | ХМ | ЛПОНП | ЛППП | ЛПНП | ЛПВП |
| Состав, % белки ФЛ ХС ЭХС ТАГ | |||||
| Функции | Перенос экзоген-ных липидов | Перенос эндоген-ных липидов | Предшест-венник ЛПНП | Перенос ХС в ткани | Перенос ХС из тканей, донор апопротеинов А, С-II |
| Место синтеза | Кишечник | Печень | Кровь | Кровь | Печень |
| Диаметр, нм | > 120 | 30-100 | 21-100 | 7-15 | |
| Основные аполипо - протеины | В-48 С-II Е | В-100 С-II Е | В-100 Е | В-100 | А-I С-II Е |
При экзогенном транспорте ресинтезированные в энтероцитах ТАГ вместе с фосфолипидами, холестеролом и белками образуют ХМ, и в таком виде секретируются сначала в лимфу, а затем попадают в кровь. В лимфе и крови с ЛПВП на ХМ переносятся апопротеины Е (апо Е) и С-II (апо С-II), таким образом ХМ превращаются в «зрелые». ХМ имеют довольно большой размер, поэтому после приема жирной пищи они придают плазме крови опалесцирующий, похожий на молоко, вид. Попадая в систему кровообращения, ХМ быстро подвергаются катаболизму, и исчезают в течение нескольких часов. Время разрушения ХМ зависит от гидролиза ТАГ под действием липопротеинлипазы (ЛПЛ). Этот фермент синтезируется и секретируется жировой и мышечной тканями, клетками молочных желез. Секретируемая ЛПЛ связывается с поверхностью эндотелиальных клеток капилляров тех тканей, где она синтезировалась. Регуляция секреции имеет тканевую специфичность. В жировой ткани синтез ЛПЛ стимулируется инсулином. Тем самым обеспечивается поступление жирных кислот для синтеза и хранения в виде ТАГ. При сахарном диабете, когда отмечается дефицит инсулина, уровень ЛПЛ снижается. В результате в крови накапливается большое количество ЛП. В мышцах, где ЛПЛ участвует в поставке жирных кислот для окисления между приемами пищи, инсулин подавляет образование этого фермента.
На поверхности ХМ различают 2 фактора, необходимых для активности ЛПЛ – апоС-II и фосфолипиды. АпоС-II активирует этот фермент, а фосфолипиды участвуют в связывании фермента с поверхностью ХМ. В результате действия ЛПЛ на молекулы ТАГ образуются жирные кислоты и глицерол. Основная масса жирных кислот проникает в ткани, где может депонироваться в виде ТАГ (жировая ткань) или использоваться в качестве источника энергии (мышцы). Глицерол транспортируется кровью в печень, где в абсорбтивный период может быть использован для синтеза жиров.
В результате действия ЛПЛ количество нейтральных жиров в ХМ снижается на 90%, уменьшаютя размеры частиц, апоС-II переносится обратно на ЛПВП. Образовавшиеся частицы называются остаточными ХМ (ремнантами). Они содержат ФЛ, ХС, жирорастворимые витамины, апоВ-48 и апоЕ. Остаточные ХМ захватываются гепатоцитами, которые имеют рецепторы, взаимодействующие с этими апопротеинами. Под действием ферментов лизосом белки и липиды гидролизуются, а затем утилизируются. Жирорастворимые витамины и экзогенный ХС используются в печени или транспортируются в другие органы.
При эндогенном транспорте ресинтезированные в печени ТАГ и ФЛ включаются в состав ЛПОНП, куда входят апоВ100 и апоС. ЛПОНП представляют собой основную транспортную форму для эндогенных ТАГ. Попав в кровь, ЛПОНП получают апоС-II и апоЕ от ЛПВП и подвергаются действию ЛПЛ. В ходе этого процесса ЛПОНП сначала превращаются в ЛППП, а затем в ЛПНП. Основным липидом ЛПНП становится ХС, который в их составе переносится к клеткам всех тканей. Образовавшиеся в ходе гидролиза жирные кислоты поступают в ткани, а глицерол кровью транспортируется в печень, где опять может использоваться для синтеза ТАГ.
Все изменения содержания ЛП в плазме крови, характеризующиеся их повышением, снижением или полным отсутствием, объединяют под названием дислипопротеинемий. Дислипопротеинемия может быть либо специфическим первичным проявлением нарушений в обмене липидов и липопротеинов, либо сопутствующим синдромом при некоторых заболеваниях внутренних органов (вторичные дислипопротеинемии). При успешном лечении основного заболевания они исчезают.
К гиполипопротеинемиям относят следующие состояния.
1. Абеталипопротеинемия возникает при редком наследственном заболевании – дефекте гена апопротеина В, когда нарушается синтез белков апоВ-100 в печени и апоВ-48 в кишечнике. В результате в клетках слизистой оболочки кишечника не формируются ХМ, а в печени – ЛПОНП, и в клетках этих органов накапливаются капельки жира.
2. Семейная гипобеталипопротеинемия: концентрация ЛП, содержащих апоВ составляет лишь 10-15% нормального уровня, но организм способен образовывать ХМ.
3. Семейная недостаточность a-ЛП (болезнь Тангира): в плазме крови практически не обнаруживаются ЛПВП, а в тканях накапливается большое количество эфиров ХС, у пациентов отсутствует апоС-II, являющийся активатором ЛПЛ, что ведет к характерному для данного состояния повышению концентрации ТАГ в плазме крови.
Среди гиперлипопротеинемий различают следующие типы.
Тип I - гиперхиломикронемия. Скорость удаления ХМ из кровотока зависит от активности ЛПЛ, присутствия ЛПВП, поставляющих апопротеины С-II и Е для ХМ, активности переноса апоС-II и апоЕ на ХМ. Генетическе дефекты любого из белков, участвующих в метаболизме ХМ, приводят к развитию семейной гиперхиломикронемии – накоплению ХМ в крови. Заболевание проявляется в раннем детстве, характеризуется гепатоспленомегалией, панкреатитом, абдоминальными болями. Как вторичный признак наблюдается у больных сахарным диабетом, нефротическим синдромом, гипотиреозом, а также при злоупотреблении алкоголем. Лечение: диета с низким содержанием липидов (до 30 г/сут) и высоким содержанием углеводов.
Тип II – семейная гиперхолестеролемия (гипер-b-липопротеинемия). Этот тип делят на 2 подтипа: IIа, характеризующийся высоким содержанием в крови ЛПНП, и IIб – с повышенным уровнем как ЛПНП, так и ЛПОНП. Заболевание связано с нарушением рецепции и катаболизма ЛПНП (дефект клеточных рецепторов для ЛПНП или изменение структуры ЛПНП), сопровождается усилением биосинтеза холестерола, апо-В и ЛПНП. Это наиболее серьезная патология в обмене ЛП: степень риска развития ИБС у пациентов с этим типом нарушения возрастает в 10-20 раз по сравнению со здоровыми лицами. Как вторичное явление гиперлипопротеинемия II типа может развиваться при гипотиреозе, нефротическом синдроме. Лечение: диета с низким содержанием холестерола и насыщенных жиров.
Тип III – дис-b-липопротеинемия (широкополосная беталипопротенемия) обусловлена аномальным составом ЛПОНП. Они обогащены свободным ХС и дефектным апо-Е, тормозящим активность печеночной ТАГ-липазы. Это ведет к нарушениям катаболизма ХМ и ЛПОНП. Заболевание проявляется в возрасте 30-50 лет. Состояние характерируется высоким содержанием остатков ЛПОНП, гиперхолестеролемией и триацилглицеролемией, наблюдаются ксантомы, атеросклеротические поражения периферических и коронарных сосудов. Лечение: диетотерапия, направленная на снижение веса.
Тип IV – гиперпре-b-липопротеинемия (гипертриацилглицеролемия). Первичный вариант обусловлен уменьшением активности ЛПЛ, повышение уровня ТАГ в плазме крови происходит за счет фракции ЛПОНП, аккумуляции ХМ при этом не наблюдается. Встречается только у взрослых, характеризуется развитием атеросклероза сначала коронарных, затем периферических артерий. Заболевание часто сопровождается понижением толерантности к глюкозе. Как вторичное проявление встречается при панкреатите, алкоголизме. Лечение: диетотерапия, направленная на снижение веса.
Тип V – гиперпре-b-липопротеинемия с гиперхиломикронемией. При этом типе патологии изменения фракций ЛП крови носят сложный характер: повышено содержание ХМ и ЛПОНП, выраженность фракций ЛПНП и ЛПВП уменьшена. Больные часто имеют избыточную массу тела, возможно развитие гепатоспленомегалии, панкреатита, атеросклероз развивается не во всех случаях. Как вторичное явление гиперлипопротеинемия V типа может наблюдаться при инсулинзависимом сахарном диабете, гипотиреозе, панкреатите, алкоголизме, гликогенозе I типа. Лечение: диетотерапия, направленная на снижение веса, диета с невысоким содержанием углеводов и жиров.
Нарушения переваривания и всасывания липидов.Поступившие с пищей жиры, если они приняты в умеренном количестве (не более 100-150 г), усваиваются почти полностью, и при нормальном пищеварении кал содержит не более 5% жиров. Остатки жировой пищи выделяются преимущественно в виде мыл. При нарушениях переваривания и всасывания липидов наблюдается избыток липидов в кале – стеаторея (жирный стул). Различают 3 типа стеаторей.
Панкреатогенная стеаторея возникает при дефиците панкреатической липазы. Причинами такого состояния могут быть хронический панкреатит, врожденнная гипоплазия поджелудочной железы, врожденный или приобретенный дефицит панкреатической липазы, а также муковисцидоз, когда наряду с другими железами повреждается и поджелудочная. В этом случае в кале содержатся желчные пигменты, понижено содержание свободных жирных кислот и повышено ТАГ.
Гепатогенная стеаторея вызывается закупоркой желчных протоков. Это происходит при врожденной атрезии желчных путей, в результате сужения желчного протока желчными камнями, или сдавления его опухолью, развивающейся в окружающих тканях. Уменьшение секреции жёлчи приводит к нарушению эмульгирования пищевых жиров, и, следовательно, к ухудшению их переваривания. В кале больных отсутствуют желчные пигменты, высоко содержание ТАГ, жирных кислот и мыл.
Энтерогенная стеаторея отмечается при интестинальной липодистрофии, амилоидозе, обширной резекции тонкого кишечника, то есть процессах, сопровождающихся снижением метаболической активности слизистой оболочки кишечника. Для этой патологии характерен сдвиг рН кала в кислую сторону, рост содержания в кале жирных кислот.
Всасывание жиров из кишечника происходит по лимфатическим путям при активной сократительной деятельности ворсинок, поэтому жировой стул может наблюдаться также при нарушении лимфооттока в случае паралича tunicae muscularis mucosae, а также при туберкулезе и опухолях мезентериальных лимфатических узлов, находящихся на пути оттока лимфы. Ускоренное продвижение пищевого химуса по тонкому кишечнику также может быть причиной нарушения всасывания жира.
ГЛАВА 20
ОБМЕН ТРИАЦИЛГЛИЦЕРОЛОВ И ЖИРНЫХ КИСЛОТ
Приём пищи человеком происходит иногда со значительными интервалами, поэтому в организме выработались механизмы депонирования энергии. ТАГ (нейтральные жиры) – наиболее выгодная и основная форма депонирования энергии. Депонированный жир может обеспечивать организм энергией при голодании в течение длительного времени (до 7-8 недель). Синтез ТАГ происходит в абсорбтивный период в печени и жировой ткани. Но если жировая ткань – только место депонирования жира, то печень выполняет важную роль превращения части углеводов, поступающих с пищей, в жиры, которые затем секретируются в кровь в составе ЛПОНП и доставляются в другие ткани. Непосредственными субстратами в синтезе жиров являются ацил-КоА и глицерол-3-фосфат. Метаболический путь синтеза жиров в печени и жировой ткани одинаков, за исключением разных путей образования глицерол-3-фосфата.
Печень – основной орган, где идет синтез жирных кислот из продуктов гликолиза. В гладком эндоплазматическом ретикулюме гепатоцитов жирные кислоты активируются и сразу же используются для синтеза ТАГ, взаимодействуя с глицерол-3-фосфатом. Синтезированные жиры упаковываются в ЛПОНП и секретируются в кровь.
В жировой ткани для синтеза ТАГ используются в основном жирные кислоты, освободившиеся при гидролизе жиров ХМ и ЛПОНП. Жирные кислоты поступают в адипоциты, превращаются в производные КоА и взаимодействуют с глицерол-3-фосфатом. Кроме жирных кислот, поступающих в адипоциты из крови, в этих клетках идет и синтез жирных кислот из продуктов распада глюкозы. Молекулы ТАГ в адипоцитах объединяются в крупные жировые капли, не содержащие воды, и поэтому являются наиболее компактной формой хранения топливных молекул.
Дата добавления: 2015-12-22; просмотров: 2335;