ГЛАВА 319. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ СОЕДИНИТЕЛЬНОЙ ТКАНИ
Дарвин Дж. Прокоп (Darwin J. Prockop)
Наследственные болезни соединительной ткани относятся к наиболее распространенным генетическим синдромам. К ним относят чаще всего несовершенный остеогенез, синдромы Элерса—Данло и Марфана.
Классификация этих синдромов основывается обычно на результатах работы McKusick, который проанализировал признаки, симптомы и морфологические изменения у большого числа больных. Однако классификация осложняется гетерогенностью этих синдромов. У больных, членов некоторых семей, отсутствует, например, один или несколько кардинальных признаков. В других семьях выявляют больных с двумя или тремя разными синдромами. Гетерогенность может быть обнаружена и среди членов одной семьи. Например, у одних больных в семье определяется дислокация суставов, характерная для синдрома Элерса—Данло, у других — хрупкость костей, типичная для несовершенного остеогенеза, а у третьих с тем же генным дефектом симптомы вообще отсутствуют. Из-за этих трудностей классификация, основанная на клинических данных, в конце концов, должна будет смениться классификацией, основанной на результатах анализа молекулярных дефектов в отдельных генах.
Организация и химический состав соединительной ткани. Соединительная ткань (или ткани) имеет довольно расплывчатое определение: внеклеточные компоненты, служащие опорой и связывающие воедино клетки, органы и ткани. К соединительным тканям относятся в основном кости, кожа, сухожилия, связки и хрящи. Они включают в себя такие кровеносные сосуды и синовиальные пространства и жидкости. На самом деле, соединительная ткань входит в состав всех органов и тканей в виде мембран и перегородок.
Соединительные ткани содержат большие количества жидкости в виде фильтрата крови, в котором находится почти половина всего альбумина организма. Большинство соединительных тканей заполнены или окружены фибриллами или волокнами коллагена (табл. 319-1) и содержат протеогликаны.
Различия соединительных тканей до некоторой степени обусловлены незначительной вариабельностью размеров и ориентации коллагеновых фибрилл. В сухожилиях они собраны в толстые параллельные пучки, в коже расположены менее упорядочение. В костях фибриллы строго организуются вокруг гаверсовых каналов, ригидность этой архитектуре придает гидроксиапатит. Основной коллаген сухожилий, кожи и костей (коллаген I типа) состоит из двух полипептидных цепей, продуктов разных структурных генов. Различия между перечисленными тканями в большой мере связаны с разной экспрессией структурных генов коллагена I типа, т. е. с разными количеством синтезируемого коллагена, толщиной и длиной образующихся фибрилл и их расположением.
Некоторые различия между соединительными тканями обусловлены присутствием ткане- или органоспецифических генных продуктов. Кости содержат белки, играющие важнейшую роль в минерализации коллагена, аорта — эластин и сопутствующий микрофибриллярный белок, несколько типов коллагена и другие компоненты. Базальная мембрана, лежащая под всеми эпителиальными и эндотелиальными клетками, содержит коллаген IV типа и другие тканеспецифические макромолекулы, а кожа и некоторые другие соединительные ткани—небольшие количества особых видов коллагена.
Таблица 319-1. Состав соединительной ткани в разных органах
| Орган | Известные компоненты | Примерное количество, % сухой массы | Свойства |
| Кожа (дерма), связки, сухожилия | Коллаген I типа | Пучки волокон с высоким пределом прочности при растяжении | |
| Коллаген III типа | 5—15 | Тонкие фибриллы | |
| Коллаген IV типа, ламинин, энтактин, нидоген | Менее 5 | В базальной мембране под эпителием и в кровеносных сосудах | |
| Коллаген V—VII типов | Менее 5 | Распределение и функции неясны | |
| Фибронектин | Менее 5 | Связан с коллагеновыми волокнами и клеточной поверхностью | |
| Протеогликаны' | 0,5 | Обеспечивают упругость | |
| Гиалуронат | 0,5 | Обеспечивает упругость | |
| Кость (демине-рализован-ная) | Коллаген'1 типа | Сложная организация фибрилл | |
| Коллаген V типа | 1—2 | Функция неясна | |
| Протеогликаны | » » | ||
| Сиалопротеины | » » | ||
| Остеонектин | 2—3 | Роль в оссификации | |
| Остеокальцин | Возможная роль в оссификации | ||
| а 2-Гликопроте ин | То же | ||
| Аорта | Коллаген I типа | 20—40 | |
| Коллаген III типа | 20—40 | Тонкие фибриллы | |
| Эластин, микрофибриллярный белок | 20—40 | Аморфное вещество, эластические фибриллы | |
| Коллаген IV типа, лами | Менее 5 | В базальной мембране | |
| нин, энтактин, нидоген | |||
| Коллаген V и VI типов | Менее 2 | Функция неясна | |
| Протеогликаны | Менее 3 | Мукополисахариды, в основном хондроитинсульфат и дерматан-сульфат; гепарансульфат в базальной мембране | |
| Хрящ | Коллаген II типа | 40—50 | Тонкие фибриллы |
| Коллаген IX и Х типов | 5—25 | Возможная роль в созревании | |
| Протеогликаны | 15—20 | Обеспечивают упругость | |
| Гиалуронат | 0,5—2 | Обеспечивает упругость |
' Протеогликановые структуры изучены недостаточно. Установлено примерно пять белковых ядер, и к каждому присоединен один вид мукополисахаридов или несколько. К основным мукополисахаридам кожи и сухожилий относятся дерматансульфат и хондроитин-4-сульфат, аорты — хондроитин-4-сульфат и дерматан-сульфат, хряща — хондроитин-4-сульфат, хондроитин-6-сульфат и кератансульфат. Базальная мембрана содержит гепарансульфат.
Биосинтез соединительной ткани. Синтез соединительных тканей заключается в самосборке из молекулярных субъединиц с точными размерами, формой и поверхностными свойствами. Молекула коллагена представляет собой длинный тонкий стержень, состоящий из трех а-полипептидных цепей, скрученных в жесткую, похожую на канат структуру (рис. 319-1). Каждая -цепь состоит из простых повторяющихся аминокислотных последовательностей, в которых каждый третий остаток представлен глицином (Гли). Поскольку каждая -цепь содержит около 1000 аминокислотных остатков, ее аминокислотную последовательность можно обозначить как (-Гли-Х-У-)ззз, где Х и Y — любые аминокислоты, кроме глицина. Тот факт, что каждый третий остаток — это глицин (самая малая аминокислота), весьма важен, так как он должен входить в стерически ограниченное пространство, в котором сходятся все три нити тройной спирали. Две -цепи в коллагене I типа одинаковы и называются 1(1). Третья же имеет несколько другую аминокислотную последовательность и называется 2(1). Некоторые типы коллагена состоят из трех одинаковых -цепей. Те участки -цепей, в которых на месте Х находится пролин или на месте Y — гидроксипролин, придают жесткость всей молекуле коллагена и удерживают ее в форме тройной спирали. Гидрофобные и заряженные аминокислоты в положениях Х и Y имеют вид кластеров на поверхности молекулы и определяют способ, которым одна молекула коллагена спонтанно связывается с другими, образуя цилиндрические фигуры, характерные для каждой коллагеновой фибриллы (см. рис. 319-1).

Рис.319-1. Схематическое изображение синтеза фибриллы коллагена I типа в фибробласте.
Внутриклеточные этапы сборки молекулы проколлагена (а): гидроксилирование и гликозилирование про-а-цепей начинается вскоре после того, как их N-концы проникнут в цистерны шероховатой эндоплазматической сети, и продолжается после сближения С-пропептидов трех цепей и образования между ними дисульфидных связей. Расщепление проколлагена с образованием коллагена, самосборка коллагеновых молекул в свободно прилегающие друг к другу нити и перекрестное связывание их в фибриллы (б): отщепление пропептидов может происходить в криптах фибробласта или в некотором отдалении от клетки (воспроизведено с разрешения из Prockop and Kivinkko).
Если структура и функция молекулы коллагена достаточно просты, то ее синтез весьма сложен (см. рис. 319-1). Белок синтезируется в виде предшественника, называемого проколлагеном, масса которого примерно в 1,5 раза больше массы молекулы коллагена. Эта разница обусловлена присутствием в проколлагене дополнительных аминокислотных последовательностей как на N-, так и на С-конце. Для образования нитей коллагена необходимо действие специфической N-протеиназы, отщепляющей N-концевые пропептиды, и специфической С-протеиназы, отщепляющей С-концевые пропептиды. По мере сборки про--цепей коллагена на рибосомах эти цепи проникают в цистерны шероховатой эндоплазматической сети. Гидрофобные «сигнальные пептиды» на N-концах отщепляются, и начинается ряд дополнительных посттрансляционных реакций. Остатки пролина в позиции Y под действием специфической гидроксилазы, требующей аскорбиновой кислоты, превращаются в оксипролин. Другая гидроксилаза в присутствии аскорбиновой кислоты точно так же гидроксилирует остатки лизина в позиции Y. Необходимость аскорбиновой кислоты для действия обеих гидроксилаз, вероятно, объясняет, почему при цинге не заживают раны (см. гл. 76). Многие гидроксилизиновые остатки подвергаются дальнейшей модификации, гликолизируясь галактозой или галактозой и глюкозой. К С-концевым пропептидам каждой цепи присоединяется крупный, богатый маннозой олигосахарид. С-концевые пропептиды сближаются, и между ними образуются дисульфидные связи. Когда в каждой про--цепй окажется примерно 100 гидропролиновых остатков, белок спонтанно сворачивается, приобретая конформацию тройной спирали. Свернувшись, белок под действием N- и С-протеиназ превращается в коллаген.
Фибриллы, образованные путем самосборки коллагеновой молекулы, обладают высоким пределом прочности при растяжении, и эта прочность еще более увеличивается за счет перекрестных реакций с образованием ковалентных связей между -цепями соседних молекул. Первый этап перекрестного связывания — окисление ферментом лизиноксидазой аминогрупп в остатках лизина и гидроксилизина с образованием альдегидов; последние затем и формируют прочные ковалентные связи друг с другом.
Коллагеновые фибриллы и волокна во всех тканях, кроме костной, стабильны на протяжении почти всей жизни и распадаются только при голодании или истощении тканей. Однако фибробласты, синовиальные и другие клетки способны продуцировать коллагеназы, расщепляющие коллагеновую молекулу в точке, отстоящей от N-конца примерно на 3/4 длины молекулы, и тем самым запускают дальнейшее разрушение коллагеновых фибрилл и волокон другими протеиназами. В костях же непрерывно происходят разрушение и ресинтез коллагеновых фибрилл, что служит необходимым условием перестройки кости. Таким образом, для сборки и сохранения коллагеновых фибрилл в тканях требуется координированная экспрессия ряда генов, продукты которых необходимы для посттрансляционного формирования этих фибрилл или участвуют в метаболизме коллагена.
Сборка фибрилл коллагена I типа аналогична сборке фибрилл коллагена II типа в хряще и коллагена III типа в аорте и коже. При формировании же нефибриллярных коллагенов, таких как тип IV в базальных мембранах, не происходит отщепления глобулярных доменов на концах молекул. Сохраняясь, эти домены участвуют в самосборке мономеров в плотные сети. Волокна эластина компонуются тем же путем. Однако эластиновый мономер представляет собой одну полипептидную цепь без четкой трехмерной структуры, самообразующую аморфные эластические волокна.
Синтез протеогликанов сходен с синтезом коллагена в том отношении, что он начинается со сборки полипептидной цепи, называемой белковым ядром. В цистернах шероховатой эндоплазматической сети белковое ядро модифицируется путем присоединения остатков Сахаров и сульфата, которые образуют крупные мукополисахаридные боковые цепи. После секреции во внеклеточное пространство белковое ядро с его мукополисахаридными боковыми цепями связывается с соединяющим белком, а затем с длинноцепочечной гиалуроновой кислотой, образуя зрелый протеогликан с относительной молекулярной массой в несколько миллионов.
Построение кости следует тем же самым принципам, что и сборка других соединительных тканей (см. также гл. 335). Первый этап заключается в отложении остеоидной ткани, которая состоит в основном из коллагена I типа (см. рис. 319-1). Далее, «еще не до конца выясненным путем происходит минерализация остеоидной ткани; особые белки, такие как остеонектин, связываются со специфическими участками коллагеновых фибрилл и затем хелируют кальций, начиная минерализацию.
Значение для наследственных болезней. Наше знание химии и биохимии соединительных тканей недостаточно полно, но тем не менее позволяет понять некоторые клинические особенности наследственных болезней этих тканей. Например, понятно, почему многие из этих болезней имеют системные проявления. Поскольку весь коллаген I типа синтезируется на одних и тех же двух структурных генах, любая мутация этих генов должна экспрессироваться во всех тканях, содержащих коллаген I типа. Тканевая или органная специфичность болезни может быть объяснена двояко. Один из механизмов может заключаться в том, что болезнь вызывается мутацией гена, экспрессирующегося только в одной или двух соединительных тканях. Например, у больных с синдромом Элерса — Данло IV типа имеются мутации генов проколлагена III типа, и его проявления ограничены изменениями кожи, аорты и кишечника, т. е. тканей, богатых коллагеном III типа. Вторая причина тканевой специфичности болезней более тонка. Разные участки молекул коллагена выполняют разные биологические функции. Так, если речь идет о коллагене I типа, то отщепление N-концевых пропептидов необходимо для сборки крупных коллагеновых фибрилл и волокон в связках и сухожилиях. При неполном отщеплении N-пропептидов белок образует тонкие фибриллы. Следовательно, больные с такими мутациями генов проколлагена I типа, препятствующих эффективному отщеплению N-пропептидов, должны страдать преимущественно дислокацией бедренных и других крупных суставов. У них редко бывают переломы, поскольку формирование толстых фибрилл коллагена I типа, по-видимому, менее важно для нормальной функции костей, чем для нормальной функции суставных связок. Наоборот, у больных с мутациями, затрагивающими структуру других участков молекулы проколлагена I типа, может преобладать костная патология.
Современные данные о химии матрикса позволяют понять причины гетерогенности симптоматики и у больных с одинаковыми генными дефектами. Экспрессия гена коллагена или протеогликана зависит от координированной экспрессии генов ферментов, принимающих участие в посттрансляционной модификации этих соединений, а также от экспрессии генов других компонентов того же матрикса. В связи с этим конечное влияние этой мутации на функциональные свойства такой сложной структуры, как кость или крупный кровеносный сосуд, зависит от различий в «генетическом фоне» разных лиц, а именно от различий в экспрессии .большого семейства других генов, продукты которых влияют на ту же структуру. Клинические проявления болезни должны зависеть и от других факторов, влияющих на соединительную ткань, таких как физическая нагрузка, травмы, питание и гормональные аномалии. Следовательно, имеется широкая основа для вариабельности клинических проявлений у больных с одним и тем же дефектом.
Выявление молекулярных дефектов. Для того чтобы выявить молекулярный дефект у больного с наследственной болезнью соединительной ткани, требуются большие усилия (рис. 319-2). Одна из причин этого заключается в том, что у двух не состоящих в родственной связи больных, даже с идентичными клиническими симптомами, молекулярные дефекты различны. Вторая причина сводится к тому, что белки и протеогликаны соединительной ткани представляют собой крупные молекулы, которые трудно перевести в раствор и получить в чистом виде. Кроме того, у больных дефект определяет синтез аномального, быстро распадающегося белка. В связи с этим при анализе тканей трудно установить, какой именно генный продукт аномален. Третья причина — большие размеры генов компонентов матрикса. В случае проколлагена I типа ген про-al (1)-цепи состоит из 18 000 пар оснований, а ген про-а2(1)-цепи— из 38000 пар. Каждый из этих генов имеет примерно 50 экзонов, большинство которых сходны по структуре. С помощью доступной в настоящее время техники рекомбинантной ДНК выяснение места мутации одного или нескольких оснований — задача неимоверной трудности. Однако новые методы позволяют, вероятно, преодолеть большинство этих проблем.
Несовершенный остеогенез
Общие проявления. Термином «несовершенный остеогенез» обозначают наследственные аномалии, обусловливающие хрупкость костей (рис. 319-3). Диагноз уста
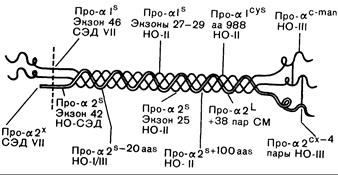
Рис. 319-2. Приблизительная локализация мутаций в структуре проколлагена I типа.
' Римскими цифрами обозначен конкретный тип синдрома Элерса—Данло (СЭД) или несовершенного остеогенеза (НО), обсуждаемых в тексте. Экзоны, в которых происходят специфические делеции, пронумерованы в направлении от 3'- к 5'-концу гена. Другие делеции обозначены примерным числом утраченных аминокислот; «аа 988» означает, что остаток глицина в положении 988 1-цепи замещен цистеином. Как сообщалось в тексте, мутация про-21 означает вставку 38 пар оснований в дополнительную последовательность и обнаружена у больных с атипичным синдромом Марфана (СМ); про-2^looaas означает делецию примерно 100 аминокислот при -варианте несовершенного остеогенеза II типа.
Про-^—мутация, ведущая к укорочению npo-al-цепи; про-(^—мутация, ведущая к укорочению ^1ро-а2-цепи; про-а!^5—мутация, ведущая к появлению цистеинового остатка; пpo-a':~ma"—мутация, ведущая к избыточному содержанию маннозы в одной или обеих про-а-цепях; про-а2" — неизвестная структурная мутация, препятствующая расщеплению цепи N-протеиназой; про-а21'— мутация, ведущая к удлинению про-а2-цепи; про-с^0" — мутация, меняющая структуру С-концевого пропептида про-а2-цепи (модифицировано и воспроизведено с разрешения из Prockop and Kivirikko).

Рис. 319-3. Мальчик в возрасте 21 мес с несовершенным остеогенезом III типа. Ребенок страдает множественными переломами рук и ног. Он гомозиготен по делеции 4 пар оснований в генах про-а2(1)-цепей, что привело к изменению последовательности последних 33 аминокислот в этих белках. В связи с этим про-а2(1)-цепи не сомкнулись с про-а1 (I) -цепями, и единственной формой проколлагенов I типа оказались тримеры про-al (I) -цепей, в которых С-концевые участки остались нескрученными (воспроизве- навливают путем исключения других наследственных дефектов или влияний факторов окружающей среды, вызывающих остеопению или остеопороз, и выявления последствий мутации в нескольких видах соединительной ткани. Повышенная ломкость костей сопровождается обычно такими признаками, как голубой цвет склер, глухота, нарушение прорезывания зубов. Эти признаки могут определяться по отдельности или вместе (табл. 319-2). Для того чтобы установить диагноз в раннем детстве, достаточно выявить сочетание голубого цвета склер и переломов. Точно так же достаточно определить сочетание переломов с характерными аномалиями зубов (несовершенный дентиногенез). Некоторые специалисты диагностическое значение придают сочетанию ломкости костей с наступившей рано глухотой у больного или членов его семьи, тогда как другие ставят диагноз только на основании хрупкости костей, которую нельзя связать с внешними факторами (такие, как малая физическая активность или сниженное питание) или с другими наследственными синдромами, например дисплазиями скелета (табл. 319-3). Поскольку у некоторых членов семей переломов не бывает до наступления постменопаузы, легкие формы болезни могут быть неотличимы от постменопаузального остеопороза. Некоторые лица с остеопорозом могут быть гетерозиготными носителями генных дефектов, вызывающих у гомозигот несовершенный остеогенез. В связи с этим целесообразно отнести постменопаузальный остеопороз в спектр тех же болезней, к которым относится несовершенный остеогенез.
Для классификации несовершенного остеогенеза пользуются классификацией, предложенной Sillence (см. табл. 319-2). Тип I встречается с частотой примерно 1:30 000. Он представляет собой легкую или средней тяжести болезнь, наследуемую как аутосомный доминантный признак в сочетании с голубыми склерами. Наиболее тяжело протекает болезнь II типа. Типы III и IV по тяжести занимают промежуточное положение между типами I и II.
Аномалии скелета. При I типе болезни ломкость костей может быть столы выраженной, что ограничивает физическую активность больного, или столь незначительной, что больной вообще не испытывает никаких неудобств. При II типе кости и другие виды соединительной ткани настолько хрупки, что смерть наступает еще в утробном периоде, в родах или в первые несколько недель после рождения ребенка. При болезни III и IV типов множественные переломы, возникающие даже при минимальных физических воздействиях, могут привести к остановке роста и костным уродствам. У многих больных переломы особенно часто возникают в детстве; после периода пубертата их частота уменьшается, а при беременности и после наступления менопаузы вновь увеличивается. Резкий кифосколиоз может быть причиной нарушений дыхания и предрасполагать к легочным инфекциям. Плотность костей снижена, но относительно специфических морфологических нарушений мнения расходятся. Общее впечатление таково, что заживление переломов происходит нормально. У некоторых больных со сравнительно легкой симптоматикой череп имеет множество вмятин, по-видимому, из-за небольших очажков оссификации.
Таблица 319-2. Классификация несовершенного остеогенеза, основанная на клинических проявлениях и способе наследования (по Sillence)
| Тип | Ломкость костей | Голубые склеры | Аномалии зубов | Глухота | Наследование |
| I | Легкая степень | Определяются | Отсутствуют при IA, определяются при 1Б | В некоторых случаях | АД |
| II | Резко выраженная | То же | В некоторых случаях | Неизвестно | AD или f Л. Г НЛп ^- |
| III | Выраженная | Голубоватый цвет при рождении | То же | Редко | AD Л.Г |
| IV | Вариабельная | Не определяются | Отсутствуют при IVA, выявляются при ГУБ | То же | АД |
Примечание. АД — аутосомное доминантное; АР — аутосомное рецессивное; С — спорадическое.
Таблица 319-3. Частичная дифференциальная диагностика несовершенного остеогенеза
| Возраст При рождении | Диагноз Гипофосфатазия | Отличительные признаки Отсутствие минерализации костей черепа |
| Ахондрогенез | Отсутствие минерализации позвонков | |
| Танатоформная карликовость Вызывающая асфиксию дистрофия грудной клетки | Н-образные позвонки Цилиндрическая форма грудной клетки | |
| Ахондроплазия | Крупная голова, короткие трубчатые кости | |
| Младенчество | Синдром детских ушибов | Чаще переломы костей черепа и ребер |
| Цинга Врожденный сифилис | ||
| Детство | Идиопатический ювенильный остеогенез | В препубертатный период, спонтанно купирующийся |
| Гомоцистинурия | Марфаноидный внешний вид и отсталость психического развития | |
| Детская диарея Опухоль коры надпочечников Лечение кортикостероидами | Стеаторея, анемия |
Источник: модифицировано из Smith et al., с. 126.
Глазные симптомы. Цвет склер варьирует от нормального до слегка голубоватого или от синевато-серого до ярко-голубого. Голубизна обусловлена истончением или прозрачностью коллагеновых волокон склеры, через которые просвечивает сосудистая оболочка глаза. У ряда больных выявляют и другие глазные симптомы. В некоторых семьях голубые склеры могут быть наследственным признаком без всякого увеличения хрупкости костей.
Несовершенный дентиногенез. Эмаль твердой зубной пластинки относительно нормальна, но зубы имеют янтарный, желтовато-коричневый или полупрозрачный голубовато-серый цвет из-за неправильного отложения дентина. Молочные зубы обычно мельче нормальных, а постоянные заострены и как бы имеют основание. Точно такие же аномалии зубов могут наследоваться независимо от несовершенного остеогенеза.
Глухота. В возрасте после 10 лет или позднее развивается глухота. Она обусловлена нарушением прохождения колебаний через среднее ухо на уровне основания стремени. При гистологическом исследовании обнаруживают недостаточную оссификацию, персистенцию хрящевых участков, которые в норме оссифицируются, и полоски скопления кальция.
Сопутствующие проявления. У многих больных и у членов многих семей выявляют аномалии и в других видах соединительной ткани. В некоторых случаях отмечают изменения кожи и суставов, неотличимые , от таковых при синдроме Элерса—Данло (см. далее). У небольшого числа больных выявляют нарушение функции сердечно-сосудистой системы, например регургитацию аортальных клапанов, пролабирование митральных, митральную недостаточность и хрупкость стенок крупных кровеносных сосудов. Могут иметь место гиперметаболизм с повышением уровня тироксина в сыворотке, гипертермия и чрезмерная потливость. При легких формах болезни сопутствующие симптомы могут выступать на первый план.
Способ наследования. Тип I болезни наследуется как аутосомный доминантный признак с непостоянной экспрессией, так что он может проявляться через поколение. При летальном варианте II типа наследование может быть аутосомным рецессивным, но в нескольких случаях II типа с выясненным генетическим дефектом имелись новые мутации. Способ наследования — основной критерий разграничения III и IV типов (см. табл. 319-2), но отличить рецессивно наследуемую форму от новой аутосомной доминантной мутации иногда очень трудно.
Молекулярные дефекты. Поскольку большинство тканей при несовершенном остеогенезе богато коллагеном I типа, считают, что многие его формы связаны с мутациями структурных генов этого белка, генов, определяющих его посттрансляционный процессинг, или генов, регулирующих его экспрессию. В настоящее время выяснены мутации генов проколлагена I типа при четырех вариантах II типа несовершенного остеогенеза. Один вариант характеризовался делецией в одном из аллелей гена про-al (I) (рис. 319-4). Она распространялась на три экзона, но не препятствовала транскрипции гена. В результате про-al (I)-цепь оказалась на 84 аминокислоты короче, чем в норме. Эта мутация была летальной, поскольку укороченная про-al (I)-цепь связывалась с нормальной про-al (I)- и про-а2(1)-цепями (см. рис. 319-4). Укорочение про-al (I)-цепи препятствовало скручиванию молекул в тройную спираль. В связи с этим большая часть проколлагеновых молекул оставалась нескрученной и быстро распадалась в процессе, называемом самоубийством белка, или негативной комплементарностью (см. рис. 319-4). При втором летальном варианте болезни II типа мутация привела к синтезу такой про-а2(1)-цепи, которая была примерно на 20 аминокислот короче по сравнению с нормой. Второй аллель не функционировал, поэтому все про-а2-цепи оказались укороченными. При третьем варианте II типа мутационная делеция в аллеле про-а2(1)-цепи укоротила синтезируемую про-а2-цепь примерно на 100 аминокислот. При четвертом варианте II типа происходило замещение одного основания, что привело к появлению в a 1(1)-цепи остатка цистеина вместо глицина и тем самым к разрыву трехспиральной конформации белка.
Мутации генов проколлагена I типа выяснены также при двух вариантах болезни III типа. При одном из них была определена делеция четырех пар оснований, что изменило последовательность последних 33 аминокислот в про-а2(1)-цепи. Больной был гомозиготен по этому дефекту, и ни одна из про-а2(1)-цепей не включалась в молекулы проколлагена. Вместо этого проколлаген I типа состоял из тримера про-al (I)-цепей. Этот тример имел трехспиральную конфигурацию, но был нестабильным. Родители больного, находившиеся друг с другом в троюродном родстве, были гетерозиготами по той же мутации и уже в возрасте 30 лет страдали остеопорозом. При другом варианте III типа структурные изменения в С-концевом пропептиде обусловили увеличение количества в нем маннозы. У больного с некоторыми симптомами болезни I типа и другими, типичными для болезни II типа, про-а2(1)-цепи были укорочены примерно на 100 аминокислот.
На основании этих данных можно сделать ряд обобщений в отношении мутаций генов коллагена. Одно из них сводится к тому, что мутация, ведущая к синтезу аномального белка, может быть более вредной, чем нефункционирующий аллель. Второе заключается в том, что мутации, обусловливающие укорочение полипептидных цепей, могут быть более частыми, чем другие. Однако у большинства больных молекулярные дефекты не идентифицированы. У многих из них могли иметь место мутации сплайсинга РНК или мутации по единичным основаниям, которые трудно обнаружить в столь крупных генах, как ген проколлагена I типа. Ряд вариантов несовершенного остеогенеза мог бы обусловливаться мутациями других генов, экспрессия которых необходима для сборки и сохранения структуры костей и других видов соединительной ткани.
Диагностика. В отсутствие кардинальных признаков болезни диагноз установить трудно, и многие случаи, вероятно, остаются недиагностированными. Следует учитывать возможность других патологических состояний, сопровождающихся хрупкостью костей в младенчестве и детстве (см. табл. 319-3). У 1/3 больных при электрофорезе проколлагена I типа (синтезируемый фибробластами кожи в культуре) в полиакриламидном геле можно обнаружить аномальную про--цепь. В большинстве случаев изменение подвижности отражает посттрансляционную модификацию и не позволяет определить точную природу мутации или тип болезни.
Лечение. Убедительные данные о возможности эффективного лечения отсутствуют. При легкой форме после уменьшения частоты переломов в возрасте 15— 20 лет больные могут и не нуждаться в лечении, но во время беременности или после наступления менопаузы, когда частота переломов снова увеличивается, к ним требуется особое внимание. При более тяжелых формах детям необходимы широкая программа физиотерапии, хирургическое лечение при переломах и. деформациях скелета, профессиональное обучение и эмоциональная поддержка как больному, так и его родителям. У многих больных интеллект достаточно развит, и они, несмотря на выраженные деформации, делают успешную карьеру. Целесообразно использовать программу поддержания позы, разработанную Bleck. При многих переломах лишь минимально смещаются кости и происходит некоторый отек мягких тканей, поэтому требуется лишь слабое вытяжение в течение 1—2 нед с последующим наложением легкой шины. При малоболезненных переломах необходимо рано начинать физиотерапию. В отношении целесообразности коррекции деформаций конечностей с помощью стального гвоздя, помещаемого в длинные кости, мнения противоречивы. Оправданием этой процедуры может служить то обстоятельство, что коррекция деформаций в детстве дает возможность взрослым больным нормально ходить.

Рис. 319-4. Схематическое изображение молекулярного дефекта при несовершенном остеогенезе II типа. а: схематическое изображение генной делеции. Как упоминалось в тексте, у человека ген про-а1(1) состоит из 18000 пар оснований и содержит около 50 экзонов (вертикальные темные черточки). Делеция захватила три экзона, содержащих 252 пары оснований кодирующих последовательностей, б: схема «самоубийства белка», или негативной комплементарности. Синтезированные укороченные про-al (1)-цепи соединились и связались дисульфидными мостиками с интактными npo-a(I)-цепями. Молекулы проколлагена, содержащие одну или две укороченные про-al (I)-цепи, не скручивались в тройную спираль при 37 °С и разрушались. В результате при спорадическом гомозиготном дефекте количество функционирующего проколлагена было уменьшено примерно на 75 % (модифицировано и воспроизведено с разрешения из Prockop and Kivirikko).
Генетическое консультирование при II, III и IV типах болезни затруднено из-за неясности способа наследования. С помощью рентгене- и эхографии несовершенный остеогенез удавалось диагностировать у плода уже на 20-й неделе беременности. В тех немногих семьях, где точно выяснен генный дефект, для пренатальной диагностики можно было бы производить анализ ДНК в соответствующих лабораториях. Для генов проколлагена I типа идентифицирован полиморфизм длины рестрикционных фрагментов, и этот подход можно было бы использовать для пренатальной диагностики. Культура клеток амниотической жидкости синтезирует коллаген, но применять эти культуры для выявления мутаций представляется нереальным.
Дата добавления: 2015-07-22; просмотров: 1205;