Гемостаз в норме
Для успешной диагностики и лечения больных с кровотечением или тромбозом необходимо знать патофизиологию гемостаза. Этот процесс можно подразделить на первичный и вторичный компоненты, а его развитие связано с травмой, хирургическим вмешательством или деструкцией выстилки сосудистого эндотелия, в результате чего кровь соприкасается с субэндотелиальной соединительной тканью. Первичный гемостаз заключается в быстром (в течение нескольких минут) формировании тромбоцитарных сгустков в месте повреждения сосуда, что имеет первоочередное значение для прекращения кровотечения из капилляров, мелких артериол и венул (см. рис. 54-1). Вторичный гемостаз, или образование фибрина, обусловлен реакциями системы'коагуляции плазмы, для завершения которых требуется более продолжительное время. В процессе этих реакций фибриновые нити скрепляют агрегаты тромбоцитов, образовавшиеся при первичном гемостазе. Это имеет особое значение для предотвращения вторичного кровотечения из крупных сосудов, наступающего через несколько часов или дней после травмы. Несмотря на различие этих процессов, первичный и вторичный компоненты гемостаза тесно связаны. Например, активированные тромбоциты, ускоренные реакции коагуляции и продукты коагуляции, такие как тромбин, стимулируют агрегацию тромбоцитов.
Эффективный первичный гемостаз представлен тремя основными этапами: 1) адгезия тромбоцитов; 2) высвобождение гранул; 3) агрегация тромбоцитов. В течение нескольких секунд тромбоциты адгезируют на нитях коллагена в сосудистом субэндотелии. Это взаимодействие опосредуется фактором Виллебранда, адгезивный гликопротеин которого позволяет тромбоцитам оставаться прикрепленными к сосудистой стенке, несмотря на значительные изменения в просвете кровеносного сосуда (рис. 54-2). Именно фактор Виллебранда выполняет задачу образования связи между рецептором тромбоцита и субэндотелиальными фибриллами коллагена. Процесс адгезии тромбоцитов с последующей продукцией и секрецией медиаторов представлен на рис. 54-1.
Активация и секреция тромбоцитов, как и других клеток, регулируются изменением уровня циклических нуклеотидов, притока ионов кальция, гидролиза мембранных фосфолипидов и фосфорилированием определенных внутриклеточных белков. Соответствующие пути представлены на рис. 54-3 и 54-4. Связывание агонистов, таких как адреналин, коллаген или тромбин, поверхностными рецепторами тромбоцитов активирует два мембранных фермента: фосфолипазы С и az. Эти ферменты катализируют высвобождение арахидоновой кислоты из двух основных мембранных фосфолипидов: фосфатидилинозитола и фосфатидил-холина. Первоначально небольшое количество высвобождаемой арахидоновой кислоты превращается в тромбоксан А.» (ТХАз), который в свою очередь может активизировать фосфолипазу С. Синтез ТХАа из арахидоновой кислоты опосредован ферментом циклоксигеназой (см. рис. 54-3). Активность этого фермента ингибируется ацетилсалициловой кислотой и нестероидными противовоспалительными средствами. Подавление синтеза ТХАа служит в целом причиной кровотечения, а также основой для действия некоторых препаратов, обладающих тромболитическими свойствами.

Рис. 54-1. Схематическое изображение основных этапов первичного гемостаза. Вначале происходят адгезия тромбоцитов, их взаимодействие с сосудистым субэндотелием, в дальнейшем — их -активация и секреция (показаны некоторые продукты секреции) и наконец связывание активированных тромбоцитов в процессе их агрегации на адгезивном монослое. АДФ — аденозиндифосфат, ФР — фактор роста (производное тромбоцитов).
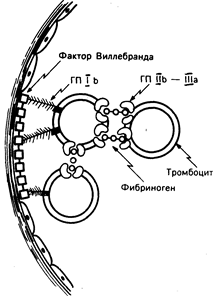
Рис. 54-2. Молекулярные основы адгезии и агрегации тромбоцитов. Адгезии тромбоцитов на сосудистом субэндотелии способствует фактор Вилленбранда, который образует мостик между нитями коллагена сосудистой стенки и рецепторами гликопротеина Ib (ГПIb) на тромбоцитах. Аналогичным образом агрегация тромбоцитов опосредуется фибриногеном, который связывает конъюгированные тромбоциты через рецепторный комплекс гликопротеина IIb и IlIa (ГПIIb— IlIa).

Рис. 54-3. Синтез тромбоксана a2 в тромбоцитах и простациклина (PGI2) в клетках эндотелия.

Рис. 54-4. Биохимические основы активации и секреции тромбоцитов. Связывание агонистов, таких как тромбин, адреналин или коллаген, с последующим запуском серии механизмов гидролиза мембранных фосфолипидов, ингибирование аденилатциклазы усиления внутриклеточного транспорта кальция и фосфорилирования жизненно важных внутриклеточных белков. В результате гранулы деформируются, движутся в направлении системы транспортных каналов, продуцируются медиаторы, подобные тромбоксану А2, и секретируются гранулы. PGI2 — простациклин, АС — аденилатциклаза, G — гуаниннуклеотидсвязывающий белок; PIP2 — фосфатидилинозитол 4,5-бифосфат, PLC—фосфолипаза С, ТХА2—тромбоксан А2, DAG — диацидглицерол, PLA2 — фосфолипаза A2, PC — фосфатидилхолин, АА — арахидоновая кислота, СО — циклоксигеназа, ip3 — инозитолтрифосфат, сАМР — циклический аденозинмонофосфат, Са-СМ — комплекс Са — модулин Са, MLCK — киназа легкой цепи миозина.
Гидролиз мембранного фосфолипида фосфатидилинозитола 4,5-бифосфата приводит к образованию диацилглицерола и инозитола трифосфата, играющего решающую роль в метаболизме тромбоцитов. Инознтола трифосфат опосредует поступление кальция в цитозол и тромбоцитов и стимулирует фосфорилирование легких цепей миозина. Последний взаимодействует с актином и тем самым способствует высвобождению гранул и изменению формы тромбоцитов. Диацилглицерол активирует протеинкиназу С, которая в свою очередь фосфорилирует протеин с относительной молекулярной массой 47000, контролирующий секрецию гранул тромбоцитов.
Известен четко сбалансированный механизм контроля за скоростью и степенью активации тромбоцитов (см. рис. 54-3). Тромбоксан А2, продукт арахидоновой кислоты тромбоцитов, усиливает активность фосфолипазы С, стимулирующей активацию и секрецию тромбоцитов. В противоположность этому простациклин (PGI2), продукт арахидоновой кислоты эндотелиальной клетки, подавляет активность фосфолипазы С путем повышения уровня внутриклеточного цАМФ, ингибирующего активацию тромбоцитов. Подобные механизмы регуляции активности и секреции происходят и в других клетках.
Вслед за активацией тромбоциты секретируют в плазму содержимое своих гранул. Эндогликозидазы и ферменты, расщепляющие гепарин, высвобождаются из лизосом, кальций, серотонин и аденозина дифосфат (АДФ) — из плотных гранул, а ряд белков, включая фактор Виллебранда, фибронектин, тромбоспондин и гепариннейтрализующий протеин (фактор IV тромбоцитов) — из -гранул. Секретируемый АДФ модифицирует поверхность тромбоцитов таким образом, что фибриноген может прикрепляться к комплексу, состоящему из мембранных гликопротеинов II b и Ilia, и связывать соседние тромбоциты с закрывающей дефект гемостатической пробкой (см. рис. 54-2). Активное участие в восстановительном процессе принимает производный тромбоцитов фактор роста, еще один протеин, синтезируемый -гранулами и стимулирующий рост и миграцию фибробластов, а также клеток гладкой мускулатуры сосудистой стенки.
В процессе формирования тромбоцитарной пробки при первичном гемостазе коагуляционные белки плазмы активируются, инициируя вторичный гемостаз. Общая схема коагуляции, включая роль разнообразных ингибиторов, представлена на рис. 54-5. Процесс коагуляции можно представить фрагментарно в виде серии реакций (рис. 54-6), заканчивающихся продукцией количества тромбина, достаточного для превращения небольшой части фибриногена плазмы в фибрин. Каждая реакция требует образования поверхностного комплекса, превращения неактивных белков-предшественников в активные протеазы путем ограниченного протеолиза и регулируется кофакторами плазмы и клеток и ионами кальция.
В процессе реакции 1, внутренней или контактной фазы коагуляции, три белка плазмы, а именно фактор Хагемана (фактор XII), высокомолекулярный кининоген и прекалликреин, образуют комплекс с коллагеном сосудистого субэндотелия. После связывания с кининогеном фактор XII медленно превращается в активную протеазу (XII а), превращающую в свою очередь прекалликреин в калликреин, а фактор XI в его активную форму XI а. Калликреин ускоряет превращение фактора XII в XII а, в то время как XI а участвует в последующей реакции коагуляции.
Реакция 2 обеспечивает второй путь коагуляции путем превращения фактора VII в активную протеазу. В этом внешнем, или зависимом от тканевого фактора, процессе образуется комплекс, между фактором VII, ионами кальция и тканевым фактором, а распределенный по всей клеточной поверхности липопротеин подвергается воздействию вслед за повреждением клетки. Фактор VII и три других белка коагуляции, а именно факторы II (протромбин), IX и X, для проявления биологической активности нуждаются в присутствии кальция и витамина К. Эти белки синтезируются в печени, в которой витамин К-зависимая карбоксилаза катализирует особую посттрансляционную модификацию путем дополнительного присоединения второй карбоксильной группы к определенным остаткам глутаминовой кислоты. Пары этих остатков ди--карбоксиглутаминовой кислоты (Gla) связывают кальций, который прикрепляет эти белки к отрицательно заряженным поверхностным фосфолипидам. Подавление этой посттрансляционной модификации синтеза белка с помощью антагонистов витамина К (например, варфарина) составляет основу большинства видов антикоагулянтной терапии.

Рис. 54-5. Диаграмма клинически важных реакций коагуляции крови. Неактивированные, или белки-предшественники обозначены римскими цифрами, а активная форма — римскими цифрами с добавлением общепринятого символа «а». Представлено два независимых пути активации: контактная и опосредованная тканевым фактором, или внешняя, системы. Обе реакции приводят к активации фактора Х и продукции тромбина, далее следует превращение фибриногена в фибрин. Регуляция этих реакций осуществляется антитромбином, образующим комплекс со всеми, относимыми к серинпротеазам, факторами, за исключением фактора VII, и системой белок С — белок S, инактивирующей факторы V и VIII.
HMWK — высокомолекулярный кининоген, РК — прекалликреин, PL — фосфолипид, ТМ — тромбомодулин.

Рис. 54-6. Основные реакции коагуляции. Они реализуются через образование комплексов поверхностных ферментов, кофакторов. РК — прекалликреин, К — калликреин, HMWK — высокомолекулярный кининоген, TF — тканевой фактор, РТ—протромбин, Т—тромбин, ломаная линия Gla (ди--карбоксиглутаминовая кислота), содержащая домены факторов VII, IX, X, Ха и РТ, которые связывают Ca2+ и фосфолипиды, штриховая линия — белки, адгезирующие на поверхности путем гидрофобного взаимодействия.
В реакции 3 фактор Х активируется протеазами, синтезированными в процессе двух предыдущих реакций. При этом образуется кальций- и липидзависимый комплекс между факторами VIII, IX и X. Внутри этого комплекса фактор IX первым превращается в IX а с помощью фактора XI а, образующегося в процессе реакции 1. Затем фактор Х активируется с -помощью фактора IX а при содействии фактора VIII. С другой стороны, фактор Х может быть активирован непосредственно фактором VII а, появляющимся в результате реакции 2. Активация фактора Х обеспечивает важную связь между внутренним и внешним механизмами коагуляции (см. рис. 54-5).
Реакция 4, завершающая процесс, заключается в превращении протромбина в тромбин в присутствии фактора V, кальция и фосфолипида. Несмотря на то что превращение протромбина может происходить на разных поверхностях, богатых фосфолипидами, оно в несколько тысяч раз усиливается на поверхности активированных тромбоцитов. Тромбин как продукт этой реакции обладает многочисленными функциями в гемостазе. Несмотря на то что его основная роль в гемостазе сводится к превращению фибриногена в фибрин, он активирует также факторы V, VIII и XIII и стимулирует агрегацию и секрецию тромбоцитов. Вслед за высвобождением фибринопептидов А и В из a- и b-цепей фибриногена модифицированная молекула, называемая мономером фибрина, полимеризуется в нерастворимый гель. Полимер фибрина при этом стабилизируется с помощью перекрестие связанных отдельных цепей фактора XIII а (плазменная трансглутаминаза).
Процесс восстановления сосудов начинается непосредственно после образования гемостатической пробки. Тканевой плазминоген-активатор (ТПА) диффундирует из эндотелиальных клеток и превращает плазминоген, адсорбированный на фибриновом сгустке, в плазмин (рис. 54-7). Затем плазмин способствует разрушению фибринового полимера на небольшие фрагменты, которые фагоцитируются и элиминируются клетками моноцитарно-макрофагальной системы. Несмотря на то что плазмин также может разрушать фибриноген, реакция остается локальной в связи с тем, что: 1) ТПА более эффективно активирует плазминоген, адсорбированный на фибриновом сгустке; 2) некоторое количество плазмина, поступающее в кровоток, быстро связывается и нейтрализуется a1-плазминовым ингибитором. Значение этого ингибитора подтверждается тем, что при его отсутствии у больного отмечается тенденция к неконтролируемым фибринолизу и кровотечениям.
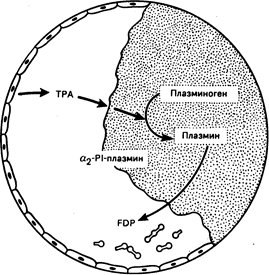
Рис. 54-7. Схематическое изображение фибринолиза.
ТРА (тканевой активатор плазминогена), отделяющийся от клеток эндотелия, представляет собой часть фибринового сгустка и активирует плазминоген в плазмин. Некоторое количество свободного плазмина образует комплекс с 2PI (ингибитор 2-плазмина). Фибрин распадается на низкомолекулярные фрагменты (FDP).
Как уже было упомянуто, система коагуляции плазмы четко регулируется таким образом, что только небольшое количество каждого фермента коагуляции превращается в активную форму, а гемостатическая пробка ограничивается локализацией дефекта. Значение регуляции велико, поскольку 1 мл крови обладает достаточным потенциалом, чтобы коагулировать весь фибриноген в организме в течение 10—15 с. Жидкое состояние крови поддерживается кровотоком как таковым, в связи с чем уменьшается концентрация реактантов, адсорбцией факторов коагуляции на поверхности клеток и активностью многочисленных ингибиторов плазмы. Антитромбин, а также белки С и S — основные ингибиторы, помогающие регулировать реакции коагуляции.
Эти ингибиторы обладают разными механизмами действия. Антитромбин формирует комплексы со всеми, относимыми к серин-протеазам, факторами коагуляции, за исключением фактора VII (см. рис. 54-5). Скорость образования комплекса увеличивается под влиянием гепарина и гепаринподобных молекул на поверхности эндотелиальных клеток. Способность гепарина усиливать активность антитромбина служит основой его антикоагулянтного действия. Белок С превращается в активную протеазу с помощью тромбина после его связывания на эндотелиальной клетке с белком, называемым тромбомодулином. Затем активированная форма белка С инактивирует два плазменных кофактора V и VIII, тем самым замедляя две основные реакции коагуляции. Белок С может также стимулировать высвобождение тканевого активатора плазминогена из эндотелиальных клеток. Ингибиторная функция белка С усиливается под влиянием белка S. Таким образом можно предположить, что снижение уровня антитромбина или белков С и S либо нефункционирующие молекулы приводят к состоянию гиперкоагуляции или претромбоза.
Эти биохимические механизмы коагуляции крови принято рассматривать как унифицированный процесс. В действительности же механизм свертывания крови варьирует в зависимости от локализации дефекта. Кроме того, отмечаются некоторые различия между гемостатическими пробками, образующимися в результате либо физиологической реакции на повреждение, либо патологического тромбоза. Чтобы подчеркнуть сходство, тромбоз часто описывают как коагуляцию без учета конкретного места и времени. Гемостатическая пробка или тромбы, которые образуются в венах с медленным кровотоком, богаты фибрином и эритроцитами, но содержат относительно мало тромбоцитов. Их часто называют красными тромбами, в связи с тем, что они образуются во время операций и в патологических участках. Рыхлые хвосты этих тромбов, которые обычно формируются в венах ног, могут отрываться и вызывать эмболию сосудов малого круга кровообращения. С другой стороны, сгустки, образованные в артериях в условиях ускоренного кровотока, состоят преимущественно из тромбоцитов и небольшого количества фибрина. Эти белые тромбы легко отделяются от артериальной стенки, поэтому могут вызвать эмболию в отдаленном участке и служить причиной временной или устойчивой ишемии. Это особенно типично для сосудов мозга и сетчатой оболочки и может привести к временной неврологической дисфункции (преходящее нарушение мозгового кровообращения), включая временную одностороннюю слепоту (преходящая слепота), или к инсульту. Наконец, накапливаются данные о том, что инфаркт миокарда в большинстве случаев обусловлен тромбами, образующимися в атеросклеротически измененных коронарных артериях.
Дата добавления: 2015-07-22; просмотров: 1437;