Хромосомные болезни
Хромосомные болезни этогруппа патологических состояний, обусловленных мутационными изменениями в хромосомном наборе (таблица 4).
Таблица 4
Частота встречаемости заболеваний, вызванных различными типами
анеуплоидии у человека
| Тип мутации | Синдромы | Частота среди новорожденных |
| Аутосомы | ||
| Трисомия 21 47,XX(XY)+21 | Дауна | 1/700 |
| Трисомия 13 47, XX(XY)+13 | Патау | 1/5 000 |
| Трисомия 18 47, XX(XY)+18 | Эдвардса | 1/10 000 |
| Половые хромосомы (женские) | ||
| ХО, Моносомия 45, XО | Шерешевского-Тернера | 1/500 |
| ХХХ, Трисомия 47, XXX | ХХХ-синдром | 1/700 |
| Половые хромосомы (мужские) | ||
| ХХУ 47, XXY | Клайнфельтера | 1/500 |
| ХХУУ 48, XXY | Клайнфельтера | 1/500 |
| ХУУ 47, XYY | Дубль У | 1/1 000 |
Показано, что примерно у 40% спонтанных абортов и 6% всех мертворожденных имеются хромосомные изменения. В то же время, около 6 из 1000 новорожденных имеют хромосомные нарушения, а удельный вес хромосомных болезней в группе детей с врожденными аномалиями составляет около 50%. Клинически почти все хромосомные болезни проявляются нарушением интеллектуального развития; множественными врожденными пороками. Это может быть умственное и физическое недоразвитие, пороки развития скелета, деформация черепа, микроцефалия, эпикант и мн. др.
Механизм возникновения геномных мутаций связан с патологией нарушения нормального расхождения хромосом в мейозе (анафаза-I и анафаза-II), в результате чего образуются аномальные гаметы (по количеству хромосом), после оплодотворения которых возникают гетероплоидные зиготы (рис. 18).
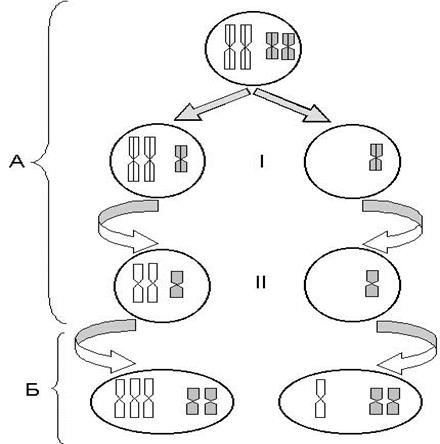
|
| Рис. 18. Схематическое изображение нерасхождения одной пары хромосом в I мейотическом делении (Н.П. Бочков и др., 1984); А – мейотическое деление I и II; Б – зиготы: 1 – трисомия, 2 – моносомия |
Хромосомные мутации (хромосомные перестройки, хромосомные аберрации) приводят к изменению числа, размеров и организации хромосом. В случае гетероплоидии особенно тяжелы моносомии. Моносомии по аутосомам заканчиваются летально еще в первые дни эмбрионального развития или приводят к гибели зародыша на более поздних стадиях (спонтанные аборты). Полные трисомии описаны у человека по большому количеству хромосом: 8, 9, 13, 14, 18, 21, X, Y. Наиболее изученными синдромами, в основе которых лежат нарушения в системе аутосом (геномные мутации, хромосомные мутации) являются трисомии 21, 13, 18, транслокационная форма Дауна, синдром «кошачьего крика», в системе половых хромосом трисомии XXY, XXX, XYY и моносомия XO.
Болезнь Дауна (трисомия 21; 47,XX(XY)+21)
Диагностика болезни Дауна уже у новорожденного не вызывает затруднений. При болезни Дауна встречается от 9 до 29 соматических аномалий. Наиболее часто при этом синдроме встречаются: брахицефальный череп со сглаженным затылком и уплощенным лицом, эпикант; пятна Брушфильда (светлые пятна на радужке); маленькие недоразвитые ушные раковины; увеличенный «складчатый» язык; широкие кисти с короткими пальцами и укороченными искривленными пятыми пальцами (клинодактилия); поперечная борозда на одной или обеих ладонях («обезьянья складка»); расширенные промежутки между 1 и 2-м пальцами стоп. Интеллектуальный дефект больных углубляется с возрастом. Известно, что примерно у 60% детей с болезнью Дауна имеются разные формы глазной патологии а у 70% обнаруживают тугоухость.
Большое внимание в последние годы уделяется изучению патогенеза синдрома Дауна. В настоящее время предложена объединенная генетическая гипотеза синдрома Дауна и болезни Альцгеймера. В статусе таких больных выявляется преждевременное старение, преобладание дегенеративных сосудистых нарушений, сахарный диабет, катаракта, липофусциноз, амилоидоз, избирательное повреждение холинергических нейронов в базальных ганглиях, склонность к злокачественным новообразованиям, специфические нарушения слуха и другие признаки, а главное – характерные нарушения интеллекта, напоминающие таковые при старческой болезни Альцгеймера.
Использование цитогенетических методов исследования показало, что примерно 80% всех случаев простой трисомии 21 имеет материнское происхождение и около 20% – отцовское. При этом лишь 20% всех случаев «материнского» синдрома Дауна обусловлено нерасхождением хромосом 21-ой пары во втором делении мейоза, а остальные – ошибками первого деления мейоза.
Болезнь Дауна транслокационной формы (46,XX(XY)t14(13,15,22)/21)
Транслокационные формы синдрома Дауна наблюдаются в 3-4% случаев. Число хромосом в данном варианте болезни нормальное – 46, так как дополнительная хромосома 21 транслоцирована на одну из аутосом (13, 14, 15 или 22) (рис.19).
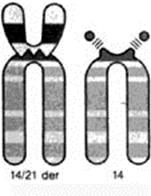
|
| Рис. 19 Транслокация 14/21. |
При этом один из фенотипически здоровых родителей является носителем сбалансированной транслокации. В кариотипе этих родителей насчитывается 45 хромосом, а одна из аутосом состоит как бы из двух частей и содержит генетический материал недостающей хромосомы, поэтому при общем числе хромосом, равном 45, нет утери генетического материала (рис. 20). Примерно в 1/3 всех случаев транслокационный вариант синдрома Дауна имеет наследственный характер. Выявление у кого-либо из родителей сбалансированной транслокации определяет необходимость пренатальной диагностики.
Синдром Эдвардса (трисомия 18; 47, XX(XY)+18 )
При кариологическом обследовании больных выявляется лишняя хромосома из группы Е (хромосома 18). Фенотипические проявления синдрома Эдвардса довольно характерны. Это наличие долихоцефального черепа, сдавленного с боков, с низким лбом и широким выступающим затылком; глазные щели узкие; эпикант; нижняя челюсть маленькая, скошена назад (микроретрогнатия); рот маленький, треугольной формы с короткой верхней губой; шея короткая, с крыловидной складкой.
При данном синдроме типичны аномалии опорно-двигательного аппарата: кисти и пальцы короткие, пятые пальцы искривлены, пальцы сжаты в кулак, второй и пятый пальцы расположены сверху и прикрывают прижатые к ладони второй и четвертый пальцы; первый палец стопы короткий и широкий, синдактилия второго и третьего пальцев; форма стопы в виде «качалки».
Почти 95% больных имеют пороки сердца, крупных сосудов, мочеполовой системы, аномалии органов пищеварения. Прогноз для жизни неблагоприятный.

Рис.20 Схема болезни Дауна транслокационной формы
Синдром Патау (трисомия 13; 47, XX(XY)+13 )
При кариологическом анализе соматических клеток больных выявляется лишняя хромосома из группы D (хромосома 13). Клиническая картина типична: микроцефальный череп с низким скошенным лбом и вдавленными височными областями; глазные щели узкие, расположены горизонтально, расстояние между ними уменьшено (гипотелоризм), почти всегда встречается глазная патология; ушные раковины расположены низко, маленькие мочки прижаты к голове, завитки неправильной формы; череп с углублениями в теменно-затылочной области, расстояние между теменными буграми увеличено.
Демонстративным признаком синдрома Патау являются «заячья губа» и «волчья пасть». Расщелины могут быть как двусторонними, так и односторонними. Почти всегда расщепление верхней губы сопровождается расщелиной неба.
Характерны также такие аномалии костно-мышечной системы, как полидактилия на верхних и нижних конечностях, второй и четвертый пальцы согнуты, приведены к ладони и перекрыты первым и пятым пальцами. Выявляются дефекты развития практически всех систем и органов. Мозг часто не разделен на полушария, наблюдается гипоплазия лобных долей, мозжечка.
У 50% больных выявляются пороки развития мочевыводящих путей: кистозная почка, гидронефроз, дисплазия почек, у 50% девочек находят удвоение влагалища и двурогую матку с гипоплазией яичников. Прогноз для жизни неблагоприятный.
Синдром «кошачьего крика» (синдром 5р–, 46XX(XY)del(5р–))
Наиболее частый из всех синдромов делеции аутосом – синдром делеции короткого плеча хромосомы 5. У больных при кариологическом анализе обнаруживается укорочение короткого плеча одной из хромосом группы В.
Фенотипическими признаками синдрома являются: микроцефалия; круглое «лунообразное» лицо в первые годы жизни и узкое лицо в более старшем возрасте; антимонголоидный разрез глаз, эпикант, косоглазие, катаракта, очаги пигментации сетчатки, атрофия зрительных нервов; плоская спинка носа, высокое небо; ушные раковины деформированы; синдактилия пальцев ног, косолапость, мышечная гипотония. Своеобразный симптом – плач при рождении, напоминающий крик кошки. Он присутствует у детей первого года жизни и обусловлен нарушением деятельности центральной нервной системы и изменениями гортани (уменьшение надгортанника, сужение гортани, отечность слизистой оболочки).
Прогноз для жизни зависит от выраженности симптомов. Многие больные доживают до подросткового возраста. Умственная отсталость всегда глубокая. Окончательный диагноз устанавливается в результате исследования кариотипа.
Синдром Шерешевского-Тернера (моносомия Х; 45, XО )
Для синдрома характерно отсутствие в кариотипе половой Х-хромосомы. Частота встречаемости 1:3000, среди девочек, страдающих олигофренией – 1:1500. Частота синдрома возрастает среди низкорослых женщин с недоразвитием вторичных половых признаков и аменореей.
Большинство больных с синдромом Шерешевского–Тернера имеют нормальный или близкий к норме интеллект, но умственная отсталость у них встречается чаще, чем в общей популяции. Интеллектуальные нарушения обычно сочетаются с недоразвитием эмоционально-волевой сферы: больные повышенно внушаемы, несколько некритичны, упрямы, часто эйфоричны.
Диагностика синдрома возможна уже в период новорожденности, так у новорожденной отмечается отечность кистей и стоп, низкий рост волос на шее, шея короткая с крыловидными складками, идущими от сосцевидных отростков к плечам. Характерна чрезмерная подвижность кожи на шее. Отмечаются множественные аномалии развития: эпикант, антимонголоидный разрез глаз; низко расположенные ушные раковины; гипомимия («лицо сфинкса»); высокое небо, аномалии зубов.
Характерны разнообразные скелетные нарушения, например «щитообразная» широкая грудная клетка, гипоплазия или сращение I и II шейных позвонков, широкие кисти с короткими IV и V пальцами, деформация локтевых и коленных суставов, укороченные III и IV пальцы стоп, синдактилия.
Важными диагностическими признаками являются также врожденные пороки сердца, низкий рост (в 98% случаев), половой инфантилизм с первичной аминореей, часты гипоплазия или гипертрофия ногтевых пластинок, гиперпигментация кожи. Наблюдаются дефекты зрения (22%) и слуха (52%).
Офтальмологическое обследование выявляет бледность сосков зрительного нерва, микрофтальм, катаракту, сужение артерий глазного дна. Дерматоглифическое исследование выявляет изменение кожных узоров пальцев и ладоней.
Диагноз может быть установлен с помощью цитологического метода исследования полового хроматина и кариологического анализа.
Синдром трисомии (47, XXX )
Для синдрома характерно наличие в кариотипе дополнительных Х-хромосом. Частота трисомии Х среди новорожденных девочек 1:800. Частота возрастает среди пациенток психиатрических больниц. В период новорожденности и детства редко можно выявить какие-либо фенотипические особенности, имеющие диагностическое значение. Основная психопатологическая особенность синдрома – проявление эмоциональной незрелости и эмоционально-поведенческие нарушения с невротическими и неврозоподобными расстройствами, иногда со склонностью к аутоагрессии. В раннем возрасте характерно выраженное отставание в развитии речи. У женщин с трисомией Х часто наблюдается эндокринный дисбаланс, бесплодие, преждевременный климакс. Могут наблюдаться более сложные полисомии Х: тетрасомия (ХХХХ) и пентасомия (ХХХХХ). Считается, что степень психического недоразвития коррелирует с числом дополнительных Х-хромосом. У женщин с полисомией Х увеличена частота психических заболеваний (шизофрения, эпилепсия, маниакально-депрессивный психоз). Окончательный диагноз устанавливается на основании цитологического обследования щечного эпителия в результате обнаружения полового хроматина в кариотипе.
Синдром Клайнфельтера (47, XXY )
Для синдрома характерно наличие в кариотипе мужчины дополнительной половой Х-хромосомы. Частота синдрома составляет в среднем 1 на 850 новорожденных мужского пола и 1–2.5% у больных олигофренией в степени дебильности. Клинические проявления достаточно вариабельны. Обязательными диагностическими критериями являются гипогенитализм и гипогонадизм. Характерными признаками также являются: высокий рост, высокое стояние таза, евнухоидные пропорции, астеническое телосложение, узкие плечи, удлиненные конечности.
Мышечная система развита слабо. Это особенно четко проявляется в препубертантном и пубертантном возрасте. У взрослых нередко встречаются склонность к ожирению по женскому типу, гинекомастия, слабое подмышечное оволосение, оволосение на лобке по женскому типу. Отмечают недоразвитие вторичных половых признаков с гипоплазией яичек и часто полового члена. Мужчины с синдромом Клайнфельтера бесплодны. Частыми являются различные диспластические признаки: брахицефалия; низкий рост волос на затылке, уплощенный затылок; гипертелоризм; эпикант; деформация ушных раковин; выступающие надбровные дуги; аномалии зубов; искривление и укорочение V пальцев.
Болезнь часто сопровождается задержкой психического развития. Диагноз может быть установлен на основании кариологического анализа, обнаружения полового хроматина в щечном эпителии.
Синдром дубль Y (47, XYY )
Синдром характеризуется наличием в кариотипе дополнительной Y-хромосомы. Наблюдается у мальчиков и мужчин высокого роста. Частота среди новорожденных мальчиков 1:840. Выраженных нарушений фенотипа может не наблюдаться. Примерно у 80% лиц с данным синдромом наблюдаются признаки психического недоразвития в сочетании с нарушениями эмоционально-волевой сферы и поведения. Больные испытывают трудности в социальной адаптации. Многим характерны замедленность и ригидность мышления, речи и моторики, часто снижена способность к самокритике. Наблюдается сочетание умственной отсталости с психопатоподобным поведением, агрессивностью, расторможенностью и извращением влечения. Отмечаются самоуверенность, импульсивность, гиперсексуальность. Окончательный диагноз устанавливается при цитологическом обследовании.
Высказывается предположение, что психопатоподобные формы поведения при наличии несбалансированного кариотипа по половым хромосомам связаны с вторичными изменениями в деятельности нервной системы как следствие нарушений гормональной сферы.
Дата добавления: 2015-07-22; просмотров: 8318;