Инсулин
Рис. 18.-1. Регуляция активности гликогенсинтазы.
Распад гликогена может проходить двумя путями.
1. Гидролитический – при участии амилазы с образованием декстринов и даже свободной глюкозы.
2. Фосфоролитический – под действием фосфорилазы и образованием глюкозо-1-фосфата. Это основной путь распада гликогена.
Фосфорилаза – сложный регуляторный фермент, существующий в двух формах – активной и неактивной. Активная форма (фосфорилаза а) – это тетрамер, в котором каждая субъединица соединена с остатком ортофосфата через гидроксильную группу серина. Под действием фосфатазы фосфорилазы происходит дефосфорилирование, отщепление 4 молекул фосфорной кислоты, и фосфорилаза а превращается в неактивную форму – фосфорилазу b, распадаясь на две димерные молекулы. Фосфорилаза b активируется путем фосфорилирования остатков серина за счет АТФ под действием фермента киназы фосфорилазы. В свою очередь этот фермент также существует в двух формах. Активная киназа фосфорилазы – фосфорилированный фермент, превращается в неактивную форму под действием фосфатазы. Активация киназы фосфорилазы осуществляется путем фосфорилирования за счет АТФ в присутствии ионов Mg2+ протеинкиназой.
Регуляция синтеза и распада гликогена носит каскадный характер и происходит путем химической модификации ферментов.
Поскольку синтез и распад гликогена протекают по разным метаболическим путям, эти процессы могут контролироваться реципрокно. Влияние гормонов на синтез и распад гликогена осуществляется путем изменения в противоположных направлениях активности двух ключевых ферментов: гликогенсинтазы и гликогенфосфорилазы с помощью их фосфорилирования и дефосфорилирования. Инсулин стимулирует синтез гликогена и тормозит распад, адреналин и глюкагон обладают противоположным эффектом.
Нарушения обмена гликогена
Гликогеновые болезни – группа наследственных нарушений в основе которых лежит снижение или отсутствие активности ферментов, катализирующих реакции синтеза или распада гликогена. К данным нарушениям относятся гликогенозы и агликогеноз.
Гликогенозы – заболевания, обусловленные дефектом ферментов участвующих в распаде гликогена. Они проявляются или необычной структурой гликогена, или его избыточным накоплением в печени, мышцах и других органах. В настоящее время предлагается деление гликогенозов на 2 группы: печеночные и мышечные.
Печеночные формы гликогенозов проявляются в нарушении использования гликогена для поддержания уровня глюкозы в крови. Общий симптом этих форм – гипогликемия в постабсорбтивный период. К этой группе относятся гликогенозы I, III, IY, YI, IX и X типов по нумерации Кори.
Мышечные формы гликогенозов характеризуются нарушениями в энергоснабжении скелетных мышц. Эти болезни проявляются при физических нагрузках и сопровождаются болями и судорогами в мышцах, слабостью и тыстрой утомляемостью. К ним относятся гликогенозы Y и YII типов.
Агликогеноз (гликогеноз О по классификации) – заболевание, возникающее в результате дефекта гликогенсинтазы. В печени и других тканях наблюдается очень низкое содержание гликогена. Это проявляется резко выраженной гипогликемией в постабсорбтивном периоде. Характерным симптомом являются судороги, особенно по утрам. Болезнь совместима с жизнью, но больные дети нуждаются в частом кормлении.
ГЛАВА 19
ЛИПИДЫ ТКАНЕЙ, ПЕРЕВАРИВАНИЕ И ТРАНСПОРТ ЛИПИДОВ
Липиды – неоднородная в химическом отношении группа веществ биологического происхождения, общим свойством которых является гидрофобность и способность растворяться в неполярных органических растворителях. Существует несколько классификаций липидов: физико-химическая, биологическая или физиологическая и структурная. Наиболее сложной является структурная классификация, основанная на структурных особенностях этих соединений. Согласно этой классификации, все липиды делятся на омыляемые и неомыляемые. К омыляемым относят те соединения, которые при щелочном гидролизе образуют соли жирных кислот (мыла), неомыляемые же липиды щелочному гидролизу не подвергаются.
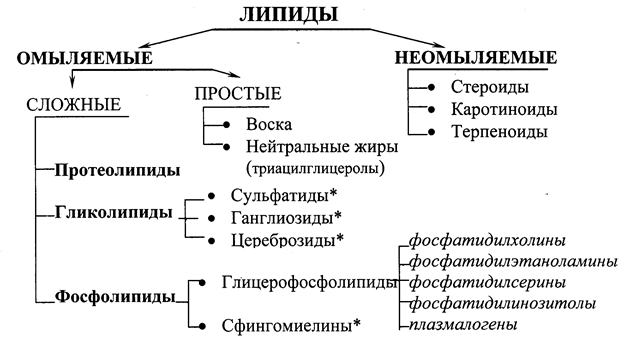
Рис. 19.1. Классификация липидов.
*В некоторых классификациях сфингомиелины, сульфатиды, ганглиозиды и цереброзиды объединяют в группу сфинголипидов, так как все они содержат аминоспирт сфингозин.
Разделение липидов по физико-химическим свойствам учитывает степень их полярности. По этому признаку липиды делятся на нейтральные или неполярные (не имеющие заряда), и полярные (несущие заряд), например, фосфолипиды и жирные кислоты. По физиологическому значению липиды делятся на резервные и структурные. Резервные липиды депонируются в больших количествах и затем расходуются для энергетических нужд организма. К резервным липидам относятся триацилглицеролы (ТАГ). Все остальные липиды можно отнести к структурным. Они не имеют особой энергетической ценности, но участвуют в построении биологических мембран и защитных покровов.
Характерным структурным компонентом большинства липидов являются жирные кислоты. Это длинноцепочечные органические кислоты, состоящие из 4-24 углеродных атомов и содержащие одну карбоксильную группу и длинный неполярный углеводородный «хвост». В составе ТАГ жирные кислоты выполняют функцию депонирования энергии. В составе фосфолипидов и сфинголипидов жирные кислоты образуют внутренний гидрофобный слой мембран, определяя его свойства. В клетках и тканях жирные кислоты встречаются в ковалентно связанной форме в составе липидов различных классов. В свободном состоянии жирные кислоты в организме содержатся в небольшом количестве, например в крови, где они транспортируются в комплексе с белком альбумином. Большинство жирных кислот образуется в организме человека, однако линолевая и линоленовая не синтезируются, поэтому обязательно должны поступать с пищей. Эти кислоты называются незаменимыми или эссенциальными. К ним относят и арахидоновую кислоту, которая может синтезироваться в организме из линолевой при достаточном поступлении последней.
Функции липидов важны и разнообразны:
- субстратно-энергетическая: жир служит в организме весьма эффективным источником энергии либо при непосредственном использовании, либо потенциально – в форме запасов жировой ткани;
- структурная (пластическая): липиды в виде комплекса с белками являются структурными элементами мембран клеток и клеточных органелл;
- транспортная: являясь одним из основных компоненнтов клеточных мембран, липиды определяют транспорт веществ в клетки;
- механическая защита: жировая прослойка предохраняет тело и органы от механических повреждений;
- теплоизолирующая: благодаря выраженной низкой термопроводимости, липиды сохраняют тепло в организме;
- электроизолирующая: липиды являются электроизолирующим материалом, участвуя таким образом в передаче нервного импульса и, соответственно, в функционировании нервной системы;
- эмульгирующая: фосфоглицеролы и желчные кислоты стабилизируют эмульсию на поверхности раздела фаз масло-вода;
- гормональная (регуляторная): стероидные гормоны, синтезируемые из холестерола, участвуют в регуляции водно-солевого обменов, половых функций; эйкозаноиды, производные полиеновых жирных кислот, вызывают разнообразные биологические эффекты;
- витаминная: в натуральных пищевых жирах содержатся жирорастворимые витамины и незаменимые жирные кислоты;
- растворяющая: одни липиды являются растворителями для других липидных веществ.
Липиды тканей человека.Липиды составляют около 10-12% массы тела человека. В среднем в теле взрослого человека содержится около 10-12 кг липидов, из них 2-3 кг приходится на структурные липиды, а остальное количество – на резервные. Основная масса резервных липидов (около 98%) сосредоточена в жировой ткани и представлена ТАГ. Эти липиды являются источником потенциальной химической энергии, доступной в периоды голодания.
Содержание липидов в тканях человека существенно различается. В жировой ткани они составляют до 75% сухого веса. В нервной ткани липидов содержится до 50% сухого веса, основные из них фосфолипиды и сфингомиелины (30%), холестерол (10%), ганглиозиды и цереброзиды (7%). В печени общее количество липидов в норме не превышает 10-14%.
Жирные кислоты, характерные для организма человека, содержат чётное число атомов углерода, чаще всего – от 16 до 20. Основной насыщенной жирной кислотой в липидах человека является пальмитиновая (до 30-35%). Ненасыщенные жирные кислоты представлены моноеновыми и полиеновыми. Двойные связи в жирных кислотах в организме человека имеют цис-конфигурацию Жиры и фосфолипиды организма при нормальной температуре тела имеют жидкую консистенцию, так как количество ненасыщенных жирных кислот преобладает над насыщенными. В фосфолипидах мембран ненасыщенных кислот может быть до 80-85%, а в составе подкожного жира – до 60%.
Липиды пищи, их переваривание и всасывание.Взрослому человеку требуется от 70 до 145 г липидов в сутки в зависимости от трудовой деятельности, пола, возраста и климатических условий. При рациональном питании жиры должны обеспечивать не более 30% от общей калорийности рациона. Жидкие жиры (масла), содержащие в своем составе незаменимые жирные кислоты, должны составлять не менее одной трети жиров пищи.
В ротовой полости и желудке взрослого человека нет ферментов и условий для переваривания липидов. Основное место расщепления липидов – тонкий кишечник. Для увеличения поверхности соприкосновения с гидрофильными ферментами жиры должны эмульгироваться (разбиться на мелкие капли). Эмульгирование происходит под действием солей желчных кислот. Эмульгированию также способствует перистальтика кишечника и выделение пузырьков СО2, происходящее при нейтрализации кислого содержимого желудка бикарбонатом, выделяющимся в составе сока поджелудочной железы.
Основная масса липидов пищи представлена ТАГ, меньше фосфолипидами (ФЛ) и стероидами. Постадийный гидролиз ТАГ осуществляется панкреатической липазой. Она секретируется в кишечник в неактивном виде и активируется колипазой и желчными кислотами. Панкреатическая липаза гидролизует жиры преимущественно в положениях 1 и 3, поэтому основными продуктами гидролиза являются глицерол, свободные жирные кислоты, моноацилглицеролы.
Фосфолипиды гидролизуются панкреатическими фосфолипазами А1, А2, С и D. Продуктами переваривания являются глицерол, жирные кислоты, фосфорная кислота и азотистые спирты (холин, этаноламин, серин, инозитол). Эфиры холестерола (ЭХЛ) расщепляются панкреатической холестеролэстеразой на холестерол (ХЛ) и жирные кислоты. Активность фермента проявляется в присутствии желчных кислот.
Всасывание липидов происходит в проксимальной части тонкого кишечника. 3-10% жиров пищи всасывается без гидролиза в виде триацилглицеролов. Основная же часть липидов всасывается лишь в виде продуктов расщепления. Всасывание гидрофильных продуктов переваривания (глицерол, жирные кислоты с числом углеродных атомов менее 12, фосфорная кислота, холин, серин, этаноламин и др.) происходит самостоятельно, а гидрофобные (ХЛ, длинноцепочечные жирные кислоты, ди- и моноглицеролы) всасываются в составе мицелл. Главную роль в образовании мицелл играют желчные кислоты. Мицелла – это сферический комплекс, в центре которого находятся транспортируемые гидрофобные продукты переваривания, окруженные желчными кислотами. Мицеллы сближаются со щеточной каймой клеток слизистой оболочки кишечника, и липидные компоненты мицелл диффундируют через мембраны внутрь клеток. Вместе с продуктами гидролиза липидов всасываются жирорастворимые витамины и соли желчных кислот. Желчные кислоты далее по воротной вене возвращаются в печень, а липидные компоненты включаются в процесс ресинтеза. В ресинтезе ТАГ участвуют не только жирные кислоты, всосавшиеся из кишечника, но и жирные кислоты, синтезированные в организме, поэтому по составу ресинтезированные жиры отличаются от полученных с пищей. Однако возможности адаптировать в процессе ресинтеза состав пищевых жиров к составу жиров организма человека ограничены, поэтому при поступлении жиров с необычными жирными кислотами в адипоцитах появляются жиры, содержащие такие кислоты. В клетках слизистой оболочки кишечника происходит синтез ФЛ, а также образование эфиров холестерола, катализируемое ацилхолестеролацилтрансферазой.
Транспорт липидов.Липиды в водной среде нерастворимы, поэтому для их транспорта в организме образуются комплексы липидов с белками – липопротеины (ЛП). Различают экзо- и эндогенный транспорт липидов. К экзогенному относят транспорт липидов, поступивших с пищей, а к эндогенному – перемещение липидов, синтезированных в организме.
Существует несколько типов ЛП, но все они имеют сходное строение – гидрофобное ядро и гидрофильный слой на поверхности. Гидрофильный слой образован белками, которые называют апопротеинами, и амфифильными молекулами липидов – фосфолипидами и холестеролом. Гидрофильные группы этих молекул обращены к водной фазе, а гидрофобные – к ядру, в котором находятся транспортируемые липиды. Апопротеины выполняют несколько функций:
· формируют структуру липопротеинов (например, В-48 – основной белок ХМ, В-100 – основной белок ЛПОНП, ЛППП, ЛПНП);
· взаимодействуют с рецепторами на поверхности клеток, определяя, какими тканями будет захватываться данный тип липопротеинов (апопротеин В-100, Е);
· являются ферментами или активаторами ферментов, действующих на липопротеины (С-II – активатор ЛП-липазы, А-I – активатор лецитин:холестеролацилтрансферазы).
Таблица 19.1.
Характеристика и состав липопротеинов
| Типы ЛП | ХМ | ЛПОНП | ЛППП | ЛПНП | ЛПВП |
| Состав, % белки ФЛ ХС ЭХС ТАГ | |||||
| Функции | Перенос экзоген-ных липидов | Перенос эндоген-ных липидов | Предшест-венник ЛПНП | Перенос ХС в ткани | Перенос ХС из тканей, донор апопротеинов А, С-II |
| Место синтеза | Кишечник | Печень | Кровь | Кровь | Печень |
| Диаметр, нм | > 120 | 30-100 | 21-100 | 7-15 | |
| Основные аполипо - протеины | В-48 С-II Е | В-100 С-II Е | В-100 Е | В-100 | А-I С-II Е |
При экзогенном транспорте ресинтезированные в энтероцитах ТАГ вместе с фосфолипидами, холестеролом и белками образуют ХМ, и в таком виде секретируются сначала в лимфу, а затем попадают в кровь. В лимфе и крови с ЛПВП на ХМ переносятся апопротеины Е (апо Е) и С-II (апо С-II), таким образом ХМ превращаются в «зрелые». ХМ имеют довольно большой размер, поэтому после приема жирной пищи они придают плазме крови опалесцирующий, похожий на молоко, вид. Попадая в систему кровообращения, ХМ быстро подвергаются катаболизму, и исчезают в течение нескольких часов. Время разрушения ХМ зависит от гидролиза ТАГ под действием липопротеинлипазы (ЛПЛ). Этот фермент синтезируется и секретируется жировой и мышечной тканями, клетками молочных желез. Секретируемая ЛПЛ связывается с поверхностью эндотелиальных клеток капилляров тех тканей, где она синтезировалась. Регуляция секреции имеет тканевую специфичность. В жировой ткани синтез ЛПЛ стимулируется инсулином. Тем самым обеспечивается поступление жирных кислот для синтеза и хранения в виде ТАГ. При сахарном диабете, когда отмечается дефицит инсулина, уровень ЛПЛ снижается. В результате в крови накапливается большое количество ЛП. В мышцах, где ЛПЛ участвует в поставке жирных кислот для окисления между приемами пищи, инсулин подавляет образование этого фермента.
На поверхности ХМ различают 2 фактора, необходимых для активности ЛПЛ – апоС-II и фосфолипиды. АпоС-II активирует этот фермент, а фосфолипиды участвуют в связывании фермента с поверхностью ХМ. В результате действия ЛПЛ на молекулы ТАГ образуются жирные кислоты и глицерол. Основная масса жирных кислот проникает в ткани, где может депонироваться в виде ТАГ (жировая ткань) или использоваться в качестве источника энергии (мышцы). Глицерол транспортируется кровью в печень, где в абсорбтивный период может быть использован для синтеза жиров.
В результате действия ЛПЛ количество нейтральных жиров в ХМ снижается на 90%, уменьшаютя размеры частиц, апоС-II переносится обратно на ЛПВП. Образовавшиеся частицы называются остаточными ХМ (ремнантами). Они содержат ФЛ, ХС, жирорастворимые витамины, апоВ-48 и апоЕ. Остаточные ХМ захватываются гепатоцитами, которые имеют рецепторы, взаимодействующие с этими апопротеинами. Под действием ферментов лизосом белки и липиды гидролизуются, а затем утилизируются. Жирорастворимые витамины и экзогенный ХС используются в печени или транспортируются в другие органы.
При эндогенном транспорте ресинтезированные в печени ТАГ и ФЛ включаются в состав ЛПОНП, куда входят апоВ100 и апоС. ЛПОНП представляют собой основную транспортную форму для эндогенных ТАГ. Попав в кровь, ЛПОНП получают апоС-II и апоЕ от ЛПВП и подвергаются действию ЛПЛ. В ходе этого процесса ЛПОНП сначала превращаются в ЛППП, а затем в ЛПНП. Основным липидом ЛПНП становится ХС, который в их составе переносится к клеткам всех тканей. Образовавшиеся в ходе гидролиза жирные кислоты поступают в ткани, а глицерол кровью транспортируется в печень, где опять может использоваться для синтеза ТАГ.
Все изменения содержания ЛП в плазме крови, характеризующиеся их повышением, снижением или полным отсутствием, объединяют под названием дислипопротеинемий. Дислипопротеинемия может быть либо специфическим первичным проявлением нарушений в обмене липидов и липопротеинов, либо сопутствующим синдромом при некоторых заболеваниях внутренних органов (вторичные дислипопротеинемии). При успешном лечении основного заболевания они исчезают.
К гиполипопротеинемиям относят следующие состояния.
1. Абеталипопротеинемия возникает при редком наследственном заболевании – дефекте гена апопротеина В, когда нарушается синтез белков апоВ-100 в печени и апоВ-48 в кишечнике. В результате в клетках слизистой оболочки кишечника не формируются ХМ, а в печени – ЛПОНП, и в клетках этих органов накапливаются капельки жира.
2. Семейная гипобеталипопротеинемия: концентрация ЛП, содержащих апоВ составляет лишь 10-15% нормального уровня, но организм способен образовывать ХМ.
3. Семейная недостаточность a-ЛП (болезнь Тангира): в плазме крови практически не обнаруживаются ЛПВП, а в тканях накапливается большое количество эфиров ХС, у пациентов отсутствует апоС-II, являющийся активатором ЛПЛ, что ведет к характерному для данного состояния повышению концентрации ТАГ в плазме крови.
Среди гиперлипопротеинемий различают следующие типы.
Тип I - гиперхиломикронемия. Скорость удаления ХМ из кровотока зависит от активности ЛПЛ, присутствия ЛПВП, поставляющих апопротеины С-II и Е для ХМ, активности переноса апоС-II и апоЕ на ХМ. Генетическе дефекты любого из белков, участвующих в метаболизме ХМ, приводят к развитию семейной гиперхиломикронемии – накоплению ХМ в крови. Заболевание проявляется в раннем детстве, характеризуется гепатоспленомегалией, панкреатитом, абдоминальными болями. Как вторичный признак наблюдается у больных сахарным диабетом, нефротическим синдромом, гипотиреозом, а также при злоупотреблении алкоголем. Лечение: диета с низким содержанием липидов (до 30 г/сут) и высоким содержанием углеводов.
Тип II – семейная гиперхолестеролемия (гипер-b-липопротеинемия). Этот тип делят на 2 подтипа: IIа, характеризующийся высоким содержанием в крови ЛПНП, и IIб – с повышенным уровнем как ЛПНП, так и ЛПОНП. Заболевание связано с нарушением рецепции и катаболизма ЛПНП (дефект клеточных рецепторов для ЛПНП или изменение структуры ЛПНП), сопровождается усилением биосинтеза холестерола, апо-В и ЛПНП. Это наиболее серьезная патология в обмене ЛП: степень риска развития ИБС у пациентов с этим типом нарушения возрастает в 10-20 раз по сравнению со здоровыми лицами. Как вторичное явление гиперлипопротеинемия II типа может развиваться при гипотиреозе, нефротическом синдроме. Лечение: диета с низким содержанием холестерола и насыщенных жиров.
Тип III – дис-b-липопротеинемия (широкополосная беталипопротенемия) обусловлена аномальным составом ЛПОНП. Они обогащены свободным ХС и дефектным апо-Е, тормозящим активность печеночной ТАГ-липазы. Это ведет к нарушениям катаболизма ХМ и ЛПОНП. Заболевание проявляется в возрасте 30-50 лет. Состояние характерируется высоким содержанием остатков ЛПОНП, гиперхолестеролемией и триацилглицеролемией, наблюдаются ксантомы, атеросклеротические поражения периферических и коронарных сосудов. Лечение: диетотерапия, направленная на снижение веса.
Тип IV – гиперпре-b-липопротеинемия (гипертриацилглицеролемия). Первичный вариант обусловлен уменьшением активности ЛПЛ, повышение уровня ТАГ в плазме крови происходит за счет фракции ЛПОНП, аккумуляции ХМ при этом не наблюдается. Встречается только у взрослых, характеризуется развитием атеросклероза сначала коронарных, затем периферических артерий. Заболевание часто сопровождается понижением толерантности к глюкозе. Как вторичное проявление встречается при панкреатите, алкоголизме. Лечение: диетотерапия, направленная на снижение веса.
Тип V – гиперпре-b-липопротеинемия с гиперхиломикронемией. При этом типе патологии изменения фракций ЛП крови носят сложный характер: повышено содержание ХМ и ЛПОНП, выраженность фракций ЛПНП и ЛПВП уменьшена. Больные часто имеют избыточную массу тела, возможно развитие гепатоспленомегалии, панкреатита, атеросклероз развивается не во всех случаях. Как вторичное явление гиперлипопротеинемия V типа может наблюдаться при инсулинзависимом сахарном диабете, гипотиреозе, панкреатите, алкоголизме, гликогенозе I типа. Лечение: диетотерапия, направленная на снижение веса, диета с невысоким содержанием углеводов и жиров.
Нарушения переваривания и всасывания липидов.Поступившие с пищей жиры, если они приняты в умеренном количестве (не более 100-150 г), усваиваются почти полностью, и при нормальном пищеварении кал содержит не более 5% жиров. Остатки жировой пищи выделяются преимущественно в виде мыл. При нарушениях переваривания и всасывания липидов наблюдается избыток липидов в кале – стеаторея (жирный стул). Различают 3 типа стеаторей.
Панкреатогенная стеаторея возникает при дефиците панкреатической липазы. Причинами такого состояния могут быть хронический панкреатит, врожденнная гипоплазия поджелудочной железы, врожденный или приобретенный дефицит панкреатической липазы, а также муковисцидоз, когда наряду с другими железами повреждается и поджелудочная. В этом случае в кале содержатся желчные пигменты, понижено содержание свободных жирных кислот и повышено ТАГ.
Гепатогенная стеаторея вызывается закупоркой желчных протоков. Это происходит при врожденной атрезии желчных путей, в результате сужения желчного протока желчными камнями, или сдавления его опухолью, развивающейся в окружающих тканях. Уменьшение секреции жёлчи приводит к нарушению эмульгирования пищевых жиров, и, следовательно, к ухудшению их переваривания. В кале больных отсутствуют желчные пигменты, высоко содержание ТАГ, жирных кислот и мыл.
Энтерогенная стеаторея отмечается при интестинальной липодистрофии, амилоидозе, обширной резекции тонкого кишечника, то есть процессах, сопровождающихся снижением метаболической активности слизистой оболочки кишечника. Для этой патологии характерен сдвиг рН кала в кислую сторону, рост содержания в кале жирных кислот.
Всасывание жиров из кишечника происходит по лимфатическим путям при активной сократительной деятельности ворсинок, поэтому жировой стул может наблюдаться также при нарушении лимфооттока в случае паралича tunicae muscularis mucosae, а также при туберкулезе и опухолях мезентериальных лимфатических узлов, находящихся на пути оттока лимфы. Ускоренное продвижение пищевого химуса по тонкому кишечнику также может быть причиной нарушения всасывания жира.
ГЛАВА 20
ОБМЕН ТРИАЦИЛГЛИЦЕРОЛОВ И ЖИРНЫХ КИСЛОТ
Приём пищи человеком происходит иногда со значительными интервалами, поэтому в организме выработались механизмы депонирования энергии. ТАГ (нейтральные жиры) – наиболее выгодная и основная форма депонирования энергии. Депонированный жир может обеспечивать организм энергией при голодании в течение длительного времени (до 7-8 недель). Синтез ТАГ происходит в абсорбтивный период в печени и жировой ткани. Но если жировая ткань – только место депонирования жира, то печень выполняет важную роль превращения части углеводов, поступающих с пищей, в жиры, которые затем секретируются в кровь в составе ЛПОНП и доставляются в другие ткани. Непосредственными субстратами в синтезе жиров являются ацил-КоА и глицерол-3-фосфат. Метаболический путь синтеза жиров в печени и жировой ткани одинаков, за исключением разных путей образования глицерол-3-фосфата.
Печень – основной орган, где идет синтез жирных кислот из продуктов гликолиза. В гладком эндоплазматическом ретикулюме гепатоцитов жирные кислоты активируются и сразу же используются для синтеза ТАГ, взаимодействуя с глицерол-3-фосфатом. Синтезированные жиры упаковываются в ЛПОНП и секретируются в кровь.
В жировой ткани для синтеза ТАГ используются в основном жирные кислоты, освободившиеся при гидролизе жиров ХМ и ЛПОНП. Жирные кислоты поступают в адипоциты, превращаются в производные КоА и взаимодействуют с глицерол-3-фосфатом. Кроме жирных кислот, поступающих в адипоциты из крови, в этих клетках идет и синтез жирных кислот из продуктов распада глюкозы. Молекулы ТАГ в адипоцитах объединяются в крупные жировые капли, не содержащие воды, и поэтому являются наиболее компактной формой хранения топливных молекул.
Дата добавления: 2015-07-18; просмотров: 1782;