Многообразные наследственные заболевания. Альбинизм
Многообразные наследственные заболевания касаются обмена тирозина. Возможно, самое известное из них — альбинизм, описанный еще античными авторами. Причиной этой болезни служат нарушения образования пигмента меланина из тирозина (см. рис. 13). Наиболее распространённая аутосомно-рецессивная форма (средняя частота заболевания 1/20 000, чаще всего встречается в Центральной Америке — 1/150, средняя частота носительства 1/70) основывается на дефекте медьсодержащего фермента меланобластов тирозиназы, которая должна превращать тирозин в 3,4-диоксифенилаланин.
Носитель полного альбинизма (альбинос или лейкопат) имеет белую кожу и волосы, розово-красные глаза. Вследствие депигментации сетчатки альбиносы, родопсин которых распадается ускоренно, плохо видят днём (дневная слепота) и им присуща фотофобия. Даже малые дозы солнечного света могут вызывать у них фотодерматит. Альбинизм поражает представителей всех рас. Генокопиями альбинизма являются его аутосомно-доминантная и сцепленая с полом формы. Аутосомно-доминантная форма проявляется чаще всего в виде частичного альбинизма.
Некоторые наследственные нарушения тирозинового обмена вызывают гибель гепатоцитов, приводящую к врождённому и раннему неонатальному циррозу.
Тирозиноз Медеса — заболевание, при котором нарушена активность пара-окси- фенилпируватдезоксигеназы. По другим данным, вовлекается дефицит митохондриальной печёночной тирозинаминотрансферазы. В отличие от алкаптонурии, при данной патологии происходит прямо противоположное — гомогентизиновая кислота в печени вообще не образуется. Это оборачивается значительно более тяжёлым метаболическим расстройством, нежели её избыток. Развиваются печёночная недостаточность и нефропатия. В оригинальном описании Дж. Медеса и соавторов (1932) больной страдал также тяжёлой миастенией, но нет достаточных оснований считать эту аутоиммунную патологию закономерным следствием тирозиноза.
Гипертпирозинемия I типа — заболевание, вызванное дефектом фумарилацетоацетат-гидролазы. В крови накапливаются тирозин и метионин. Больные гибнут во младенчестве от печёночной недостаточности и нефропатии. Аутопсия обнаруживает цирроз печени и тубулонекроз. Гипертпирозинемия II типа возникает вследствие дефицита цитоплазматической тирозинаминотрансферазы печени. Она протекает не столь фатально, но обусловливает выраженную задержку психомоторного развития.
Хоукинсурия — нарушение активности 4-гидроксифенилпируватдезоксигеназы. Она также сопровождается тирозинемией, но, в отличие от всех вышеназванных аминоацидопатий, имеет аутосомно-доминантный тип наследования. Болезнь характеризуется задержкой психомоторного развития (ЗПМР), степень которой варьирует в зависимости от глубины нарушения обмена глутатиона и формирования эпоксидных метаболитов ароматических аминокислот.
Преходящая тирозинемия с тирозинунезрелости вышеназванного печёночного медьсодержащего фермента. Так как аскорбиновая кислота повышает активность 4- гидроксифенилпируватдезоксигеназы, терапия витамином С способствует быстрейшей коррекции обмена.
Из-за того, что тирозин — сырьё для продукции тироидных гормонов, при некоторых тирозинопатиях формируется наследственный первичный гипотироз. В этом отношении заслуживает упоминания дефект йодтирозиндейодиназы. При данном аутосомно-рецессивном заболевании моно- и дийодтирозин не дейодируются и йод секвестрируется в этих гормонально неактивных метаболитах тирозина. Развивается нехватка тироидных гормонов (рис. 13) и компенсаторная гиперплазия щитовидной железы до статуса зоба.
В силу врождённого характера гипотироза нарушается развитие мозга и обучаемость, так как тироидные гормоны — стимуляторы аминоацил-т-РНК-синтетаз — существенны для синтеза быстрообмениваемых пептидов ЦНС. Отмечается кретинизм. Имеется модель болезни на кроликах с аналогичным дефектом. В изолятах, за счет эффекта родоначальника и инбридинга, частота данного дефекта возрастает, что не должно неправильно интерпретироваться, как указание на эндемичную геохимическую йодную аномалию (см. ниже «Патофизиология эндокринной регуляции»).
Большое клиническое значение имеет наследственная оксалурия — причина раннего оксалат-кальциевого нефроуролитиаза и быстропрогрессирующей хронической почечной недостаточности. При I типе этого заболевания присутствует дефект пероксисомального фермента аланин-глиоксилат-аминотрансферазы, при II типе — дефекты редуктаз глиоксиловой кислоты. В обоих вариантах аутосомно-рецессивные дефекты приводят к накоплению глиоксиловой кислоты и ее переходу в щавелевую кислоту, кальциевые соли которой формируют кристаллы в мочеполовой системе, а при I типе — и в сосудах, сердце, костях.
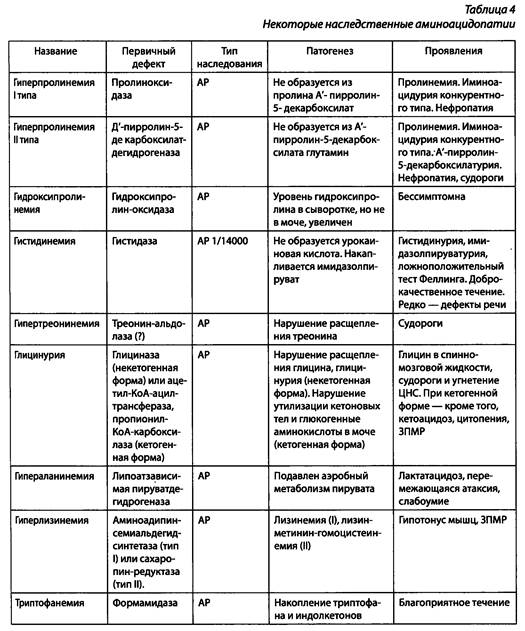
Витамин В6 способствует облегчению оксалурии. В таблице 4 можна найти краткую характеристику еще нескольких наследственных аномалий межуточного обмена аминокислот.
Исключая эндокринопатии, рассмотренные ниже, приобретённые нарушения обмена аминокислот могут быть вызваны, прежде всего, различными факторами, расстраивающими процессы переаминирования.
При авитаминозе В6 (см. также ниже «Патофизиология витаминного обмена») переаминирование и дезаминирование аминокислот в печени полностью нарушается. Такая ситуация возможна не только при недостатке витамина В6 в диете, что бывает довольно редко. Большое значение имеют нарушения, связанные с действием антагонистов трансаминаз, к которым относятся фтивазид и его аналоги и циклосерин. При терапии туберкулёза нарушение трансаминирования, вследствие побочного действия лекарств, усугубляет катаболический эффект цитокинов, образуемых при хроническом инфекционном процессе. Недостаток синтеза апоферментов-трансаминаз сопровождает голодание и печеночную недостаточность, как при развитии цирроза, так и стеатоза печени и даже при выраженном гепатите.
При хроническом алкоголизме потребность в витамине В6 растёт, а возможности трансаминирования в печени сокращаются. Вследствие этого у алкоголиков наблюдается зачастую катаболический сдвиг белкового обмена и отрицательный азотистый баланс. Как уже упоминалось выше в разделе «Общий обзор путей метаболизма аминокислот», дефицит а-кетокислот также тормозит переаминирование. Поэтому, нарушение данного процесса (и аминирования-дезаминирования) наблюдается при глубокой тканевой гипоксии, гиповитаминозе, В, и В2 и любом торможении окислительно-восстановительных ферментов цикла Кребса или ограничении его пропускной способности.
Все вышеупомянутые состояния характеризуются гипераминоацидемией, аминоацидурией и увеличенной потерей немочевинного азота с мочой.
Особое значение для диагностики имеют ситуации, когда ферменты-аминотрансферазы оказываются в увеличенном количестве в плазме крови. Это свидетельствует об усиленных процессах цитолиза в том или ином органе. Отдельные трансаминазы содержатся в различных органах в неодинаковых количествах. Больше всего аспартатаминотрансферазы (АСАТ) содержат кардиомиоциты, затем по убыванию концентраций идут печень, скелетные мышцы, почки и поджелудочная железа.
Аланинаминотрансфераза (АЛАТ) присутствует в рекордном количестве в печени, затем идут поджелудочная железа, миокард и скелетная мускулатура. Соответственно, повышение уровня АСАТ в крови считается более характерным для инфаркта миокарда, а нарастание активности АЛАТ может свидетельствовать о цитолизе гепатоцитов (А.Ф. Блюгер, 1976). Впрочем, данные тесты неспецифичны.
Приобретенные нарушения могут затрагивать и другие аспекты интермедиарного метаболизма аминокислот.
Так, при алиментарной дистрофии, подобно фенилкетонуриям, нарушается окисление фенилаланина и могут возникать те же токсические для ЦНС продукты, что и при фенилпировиноградной олигофрении, например, фенилпируват. Это заставляет предполагать, что не только «Сытое брюхо к ученью глухо», но и продолжительные голодовки не благоприятствуют максимальному проявлению умственных способностей.
Биосинтез многих заменимых аминокислот имеет этапы, зависящие от присутствия дериватов витамина никотиновой кислоты, и страдает при пеллагре. Катаболизм триптофана особенно чувствителен к витаминной недостаточности и может тормозиться на разных этапах ещё и при гиповитаминозах В1, В2, В6, а также при гиперкортицизме.
Нарушение окисления тирозина наблюдается при гипертирозе, цинге и дефиците меди.
Биосинтез серина нарушается при гипокортицизме и кастрации, а при превращении серина в глицин центральную роль играет витамин фолиевая кислота — и этот процесс страдает при соответствующем гиповитаминозе.
Большое значение в медицине имеют приобретенные нарушения переметили- рования, при котором метальные группы серина и метионина вовлекаются в метаболизм, переносясь на другие акцепторы. Метилтрансферазы, необходимые для этого, могут использовать метилированную форму тетрагидрофолиевой кислоты и витамин В12, как метальные донаторы. Таким образом, эти витамины необходимы для обмена серосодержащих аминокислот — метионина и цист(е)ина. Вместе с холином, бетаином, кобаламином метионин служит важным источником метильных групп, необходимых при синтезе ЛПОНП и ЛПВП печенью, и сам входит в состав их апопротеинов.
Метальные группы из состава метионина необходимы для формирования фосфатидилхолина, используемого при построении оболочек липопротеидов в печени. Без достаточного количества фосфатидилхолина экскреция липопротеидов печенью тормозится. Кроме того, метильные донаторы участвуют в построении триметилированного производного аминокислоты лизина — карнитина. Карнитин — челнок, транспортирующий в митохондрию для окисления остатки длинноцепочечных жирных кислот. Вполне понятно, что при его нехватке утилизация липидов печенью замедляется. Таким образом, при дефиците метионина и других вышеназванных метилдающих соединений, получивших название липотропных веществ, происходит ожирение печени. Так как при этом нарушена продукция антиатерогенных ЛПВП, можно полагать, что этим объясняется взаимосвязь между дефицитом метильных донаторов, гомоцистинурией и ускорением атеросклероза (см. выше).
Образование порфириновых соединений из аминокислот и их производных (глицина и сукцинил-КоА), необходимое для синтеза гема, контролируется ферментом 8-аминолевулинат-синтетазой, коферментом которого служит витамин В6, а активный центр содержит ион Mg2+. При гиповитаминозе В6, в частности, вызванном алкоголизмом, а также при отравлении свинцом, конкурирующим с магнием активность данного фермента нарушается, что ведет к ослаблению синтеза порфиринов и сидеробластическим анемиям. Указанный фермент столь чувствителен к РЬ2+, что его определение рекомендовано для раннего скрининга хронической свинцовой интоксикации (сатурнизма).
Дата добавления: 2022-06-14; просмотров: 4037;