Тромбоциты – кровяные пластинки.
1. Тромбоциты не могут считаться полноценными клетками, поскольку не содержат ядра.
2. В тромбоцитах протекают основные биохимические процессы: синтез белка, реакции обмена углеводов и липидов, окислительное фосфорилирование.
3. Основная функция тромбоцитов – участие в процессе свертывания кровиобусловлена наличием тромбоцитарных факторов свертывания.
Гемоглобин человека
Гемоглобин – сложный железосодержащий белок, относится к классу гемопротеинов. Выполняет две важные функции:
1. перенос кислорода из легких к периферическим тканям;
2. участие в переносе СО2 и протонов из периферических тканей в легкие.
Производные гемоглобина
Молекула гемоглобина взаимодействует с различными лигандами, образуя производные гемоглобина.
1. Дезоксигемоглобин – ННb – не связанный с кислородом и содержащий гем с двухвалетным железом Fe2+.
2. Оксигемоглобин – ННbO2 – полностью оксигенированный гемоглобин, связанный с четырьмя молекулами кислорода.
3. Карбгемоглобин – ННbCO2 – гемоглобин, связанный с СО2. Выполняет функцию выведения СО2 из тканей к легким. Соединение нестойкое, легко диссоциирует в легочных капиллярах. Этим путем выводится до 10–15% СО2.
4. Карбоксигемоглобин – ННbСО – образуется при отравлении оксидом углерода (II). Сродство гемоглобина к СО примерно в 300 раз выше, чем к кислороду, при этом гемоглобин теряет способность связывать кислород и наступает смерть от удушья.
5. Метгемоглобин – MetHb – образуется при действии окислителей (нитрит натрия, нитробензол). Содержит железо в трехвалентной форме Fe3+ и теряет способность к переносу кислорода. В норме образуется небольшое количество метгемоглобина – примерно 0,5 % в сутки.
Варианты гемоглобина в онтогенезе
Количество и состав фракций гемоглобина изменяется в процессе онтогенеза. Все гемоглобины представляют собой тетрамеры, построенные из разного набора субъединиц (α, β, γ, δ) и преимущественно образуются на разных этапах развития организма человека – от эмбрионального до взрослого состояния. Различают следующие физиологические типы гемоглобинов: примитивный гемоглобин НbР, фетальный гемоглобин HbF (fetus – плод), гемоглобин взрослых HbA, HbA2, HbA3 (adultus – взрослый).
Примитивный гемоглобин – синтезируется в эмбриональном желточном мешке через несколько недель после оплодотворения. Состоит из двух α‑ и двух ε‑цепей (2α, 2ε). Через две недели после формирования печени плода в ней начинает синтезироваться HbF, который к шести месяцам полностью замещает НbР.
Фетальный гемоглобин – синтезируется в печени и костном мозге плода до периода его рождения. Состоит из двух α‑ и двух γ‑цепей (2α, 2γ). Характеризуется более высоким сродством к кислороду и обеспечивает эффективную доставку кислорода к эмбриону из системы кровообращения матери. HbF является главным типом гемоглобина плода. Кровь новорожденного содержит до 80% HbF, но к концу 1‑го года жизни он почти целиком заменяется на HbA. В крови взрослого человека присутствует в минимальном количестве – до 1,5% от общего количества гемоглобина.
Гемоглобин А – основной гемоглобин взрослого человека (96 % от общего количества). Начинает синтезироваться в клетках костного мозга уже на 8‑м месяце развития плода. HbA состоит из двух α‑ и двух β‑цепей.
Минорные гемоглобины:
1. HbA2 ‑ 2α 2δ, в крови взрослого человека примерно 2,6 % HbA2. Обладает большим сродством к кислороду.
2. HbA3 ‑ 2α 2β, однако имеются изменения в строении β‑цепей по сравнению с HbA. Появляется в крови в небольших количествах при старении.
Гемоглобинопатии
Все структурные аномалии белковой части гемоглобина называют гемоглобинозами.
Различают:
1. гемоглобинопатии;
2. талассемии.
Гемоглобинопатии – наследственные изменения структуры какой‑либо цепи нормального гемоглобина вследствие точечных мутаций генов. Известно около 300 вариантов HbA, имеющих в первичной структуре α‑ или β‑цепи незначительные изменения. Некоторые из них практически не влияют на функции белка и здоровье человека, другие – вызывают значительные нарушения функции HbA и развитие заболеваний различной степени тяжести.
В аномальных гемоглобинах изменения могут затрагивать аминокислоты:
1. находящиеся на поверхности белка;
2. участвующие в формировании активного центра;
3. аминокислоты, замена которых нарушает трехмерную конформацию молекулы;
4. аминокислоты, замена которых изменяет четвертичную структуру белка и его регуляторные свойства.
Аномальные гемоглобины отличаются от HbA по первичной структуре, форме, величине заряда. При этом изменяются такие свойства как сродство к кислороду, растворимость, устойчивость к денатурации и др.
Примеры.
1. Серповидноклеточная анемия. Наследственное заболевание, связанное с заменой глутаминовой кислоты в 6‑м положении (с N‑конца) на валин в β‑цепях молекулы гемоглобина S. Растворимость дезоксигемоглобина S значительно снижена. Его молекулы начинают «слипаться», образуя волокнистый осадок, который деформирует эритроцит, придавая ему форму серпа (полумесяца). Такие эритроциты плохо проходят через капилляры тканей, закупоривают сосуды и создают локальную гипоксию. Они быстро разрушаются и возникает гемолитическая анемия. Дети, гомозиготные по мутантному гену, часто умирают в раннем возрасте. Болезнь распространена в странах Южной Америки, Африки и Юго‑Восточной Азии.
2. Гемоглобин М – в результате мутации в гене происходит замена в α‑ или β‑цепи гистидина (в 7‑м или 8‑м положении) на тирозин. В результате этого Fe2+ окисляется в Fe3+ и образуется метгемоглобин, не способный связывать кислород. Развивается цианоз и гипоксия тканей.
Талассемии
Талассемии – наследственные заболевания, связанные с нарушением синтеза α‑ или β‑цепей.
β‑талассемии развиваются в результате снижения синтеза β‑цепей. Проявляется после рождения, при этом в крови наряду с НbА появляется до 15 % НbА2 и 15–60 % HbF. Болезнь характеризуется гиперплазией и разрушением костного мозга, поражением печени, селезенки и сопровождается гемолитической анемией.
α‑талассемии возникают при нарушении синтеза α‑цепей. При полном отсутствии α‑цепей наступает внутриутробная гибель плода, так как не образуется HbF, а тетрамеры γ4 обладают высоким сродством к кислороду и не способны выполнять транспортную функцию, что ведет к развитию тканевой гипоксии и к смерти вскоре после рождения.
Обмен железа
В организме взрослого человека содержится 3–4 г железа, из этого количества около 3,5 г находится в плазме крови. Гемоглобин эритроцитов содержит примерно 68 % всего железа организма, ферритин – 27 % (резервное железо печени, селезенки, костного мозга), миоглобин (в мышцах) – 4 %, трансферрин (в плазме крови) – 0,1. На долю всех содержащих железо ферментов приходится примерно 1 % железа, имеющегося в организме.
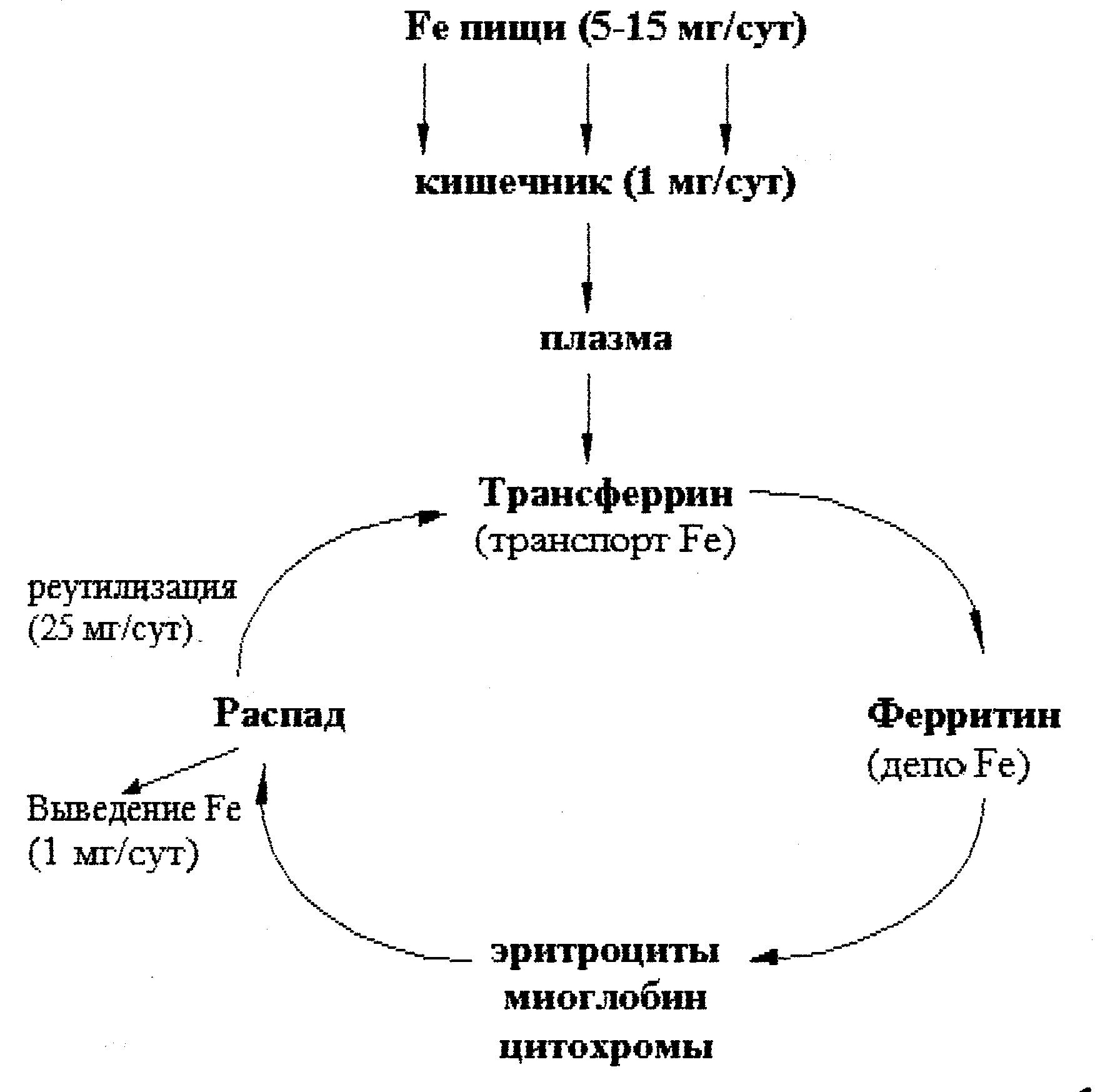
Рис. 30.1. Обмен железа в организме человека.
В обмене железа принимает участие ряд белков.
Апоферритин. Белок связывает железо в эритроцитах и превращается в ферритин, который остается в энтероцитах. Таким способом регулируется поступление железа в капилляры крови из клеток кишечника. Когда потребность организма в железе невелика, скорость синтеза апоферритина повышается. При недостатке железа в организме апоферритин в энтероцитах почти не синтезируется.
Трансферрин. Это транспортный белок, относится к гликопротеинам, синтезируется в печени. Он имеет два центра связывания железа. Трансферрин транспортирует железо с током крови к местам депонирования и использования. В норме трансферрин насыщен железом приблизительно на 33 %.
Ферритин. Олигомерный белок с молекулярной массой 450 к Да. Он состоит из 24 идентичных протомеров, образующих полую сферу. Железо депонируется в ферритине в виде гидроксифосфата. Содержание железа в молекуле ферритина непостоянно. Функция ферритина – депонирование железа. Ферритин содержится почти во всех тканях, но в наибольшем количестве в печени, селезенке, костном мозге.
Дата добавления: 2016-01-30; просмотров: 954;