Закономерности управления сложными процессами
Сложными называются процессы, в которых наряду с реакцией, приводящей к образованию целевого продукта (целевой реакцией), протекают побочные реакции.
Характеристика модели:
1) Выход целевого продукта не равен конверсии реагента β ≠ α;
2) Выход целевого продукта, даже теоретически не может достигнуть значения 100% β < 100%.
Объекты управления:
Наряду со скоростью процесса, положением равновесия (если реакция обратима) объектом управления в сложных процессах становится селективность.
r = f (С, р, t, kat) или r = f (W, ω, f) в зависимости от лимитирующей стадии
α* = f (C, p, t)
S = f (C, p, t, kat)
Инструменты управления: кинетические параметры.
Рассмотрим, как влияют основные технологические параметры на скорость и селективность сложного необратимого гомогенного процесса.
Пусть протекают две параллельные реакции, в которых расходуется реагент А. 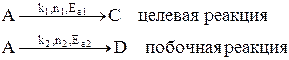
Кинетические кривые для сложно-параллельного процесса имеют вид, представленный на рисунке. Концентрация исходного реагента А уменьшается во времени, а концентрация продуктов C и D увеличивается, причем скорость увеличения зависит от порядка реакции. При n1 > n2 быстрее увеличивается концентрация целевого продукта.
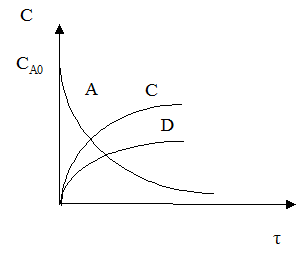
Для анализа воспользуемся понятием дифференциальной селективности, под которой понимают отношение скорости целевой реакции (скорости образования целевого продукта) к общей скорости процесса (скорости расходования реагента).
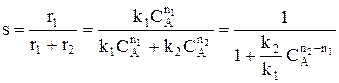
При условии, что 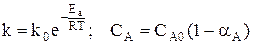
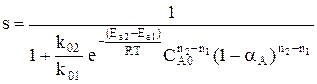
Используя полученную формулу, проведем анализ влияния начальной концентрация реагента, степени его превращения, давления и температуры на селективность процесса.
Характер влияния концентрации, давления и конверсии зависит от соотношения порядков целевой и побочной реакции.
Если порядки равны, начальная концентрация, давление и конверсия не влияют на селективность процесса. Если порядок целевой реакции выше, селективность увеличивается при повышении концентрации реагента и давления и уменьшается при достижении высоких конверсий. Если порядок целевой реакции ниже, чем порядок побочной реакции, при увеличении концентрации реагента и давления селективность должна понижаться, а увеличение времени реакции (конверсии реагента) приводит к повышению селективности.
При СА0 ↑ rхим. р. ↑ если n1 = n2 s = const
если n1 > n2 s↑
если n1 < n2 s↓
При αА↑ rхим. р.↓ если n1 = n2 s = const
если n1 > n2 s↓
если n1 < n2 s↑
Для газофазных реакций
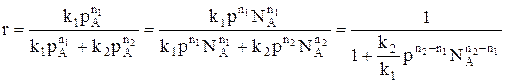
При р↑ rхим. р. ↑ если n1 = n2 s = const
если n1 > n2 s↑
если n1 < n2 s↓
Влияние температуры зависит от соотношения энергий активации целевой и побочной реакции.
При t ↑ rхим. р. ↑ если Еа1 = Еа2 s = const
если Еа1 > Еа2 s↑
если Еа1 < Еа2 s↓
Рассмотрим другой тип сложных процессов – сложно-последовательный необратимый гомогенный процесс (консекутивный).
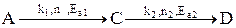
С течением времени концентрация реагента А снижается. Концентрация промежуточного продукта С вначале увеличивается, достигает своего максимального значения, затем убывает. Концентрация продукта D увеличивается по мере протекания реакции. Величина максимальной концентрации продукта С зависит от соотношения скоростей каждой из последовательных стадий.
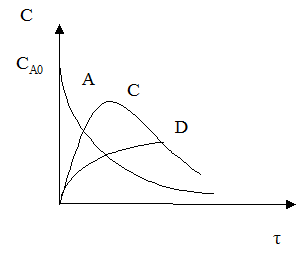
Если целевым продуктом является продукт С, то селективность процесса равна
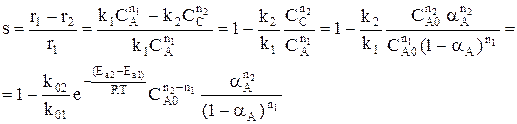
При увеличении начальной концентрации реагента
СА0 ↑ rхим. р. ↑ если n1 = n2 s = const
если n1 > n2 s↑
если n1 < n2 s↓
При увеличении степени превращения реагента
αА↑ rхим. р , s↓
При увеличении температуры
t ↑ rхим. р. ↑ если Еа1 = Еа2 s = const
если Еа1 > Еа2 s↑
если Еа1 < Еа2 s↓
На селективность газофазных процессов будет оказывать влияние давление.
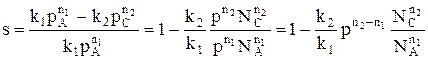
При р↑ rхим. р. ↑ если n1 = n2 s = const
если n1 > n2 s↑
если n1 < n2 s↓
Анализ показал, что в некоторых случаях изменение технологического параметра может привести к увеличению скорости процесса, а селективность при этом уменьшится или к увеличению селективности при снижении скорости процесса. Выбор оптимального значения этого технологического параметра производится путем технологических и экономических расчетов. Очень часто предпочитают получить высокую селективность, особенно если сырье имеет высокую стоимость, высока стоимость отделения и очистки целевого продукта от побочных продуктов, а скорость процесса невелика.
Обобщая вышесказанное, можно сделать следующие выводы:
1) При разработке технологии сложных процессов основное внимание следует уделить селективности процесса. Другими словами, нужно найти условия, обеспечивающие преимущественное протекание целевой реакции.
2) Условия проведения процесса определяются на основании экспериментального изучения кинетики процесса (определения порядка всех реакций, энергии активации и др.). Найденные условия можно менять в узком диапазоне.
3) Самое лучшее решение в задаче повышения селективности процесса – это нахождение селективного катализатора.
Пример Получение азотной кислоты. Стадия окисления аммиака
Азотная кислота является одним из важнейших многотоннажных продуктов химической промышленности. Процесс ее производства складывается из трех стадий:
1) окисление аммиака с целью получения оксида азота (II)
4NH3 + 5O2 → 4NО + 6H2O;
2) окисление NO до NO2 2NO + O2 ↔ 2NO2;
3) абсорбция оксидов азота водой 4NO2 + O2 + 2H2O → 4HNO3 .
Для выбора инструментов управления первой стадией процесса дадим технологическую классификацию процесса: процесс сложный, необратимый, экзотермический, каталитический, гомофазный, но гетерогенный.
При сгорании аммиака в отсутствии катализатора образуются азот и вода. 4NH3 + 3O2 → 2N2 + 6H2O + 1268 кДж/моль
Если использовать марганцевый катализатор, то при окислении аммиака с выходом 95% образуется N2O.
4NH3 + 4O2 → 2N2О + 6H2O + 1105 кДж/моль
При использовании платинового катализатора с выходом 95% образуется окись азота NO. 4NH3 + 5O2 → 4NО + 6H2O + 986 кДж/моль
Платина обладает низкой температурой зажигания (2000С), хорошую пластичность, тягучесть. Недостаток платины – ее быстрое разрушение при высоких температурах под воздействием больших скоростных потоков реагентов и каталитических ядов. Более стабильными являются модифицированные родием и палладием катализаторы. Наиболее распространены следующие катализаторы окисления аммиака:
Pt + 4%Pd +3,5%Rh (для работы при атмосферном давлении)
Pt + 7,5%Rh (для работы при повышенном давлении).
Эти катализаторы изготавливают в виде сеток из проволоки диаметром 0,09 мм, размер стороны ячейки 0,22 мм, число ячеек на 1 см длины – 32, на 1 см2 – 1024. Модифицированные платиновые катализаторы весьма чувствительны к ядам; наиболее сильными ядами являются соединения серы и фтора. Разрушают также сетки пыль и оксиды железа, попадающие в исходную смесь при синтезе аммиака; они к тому же, засоряя сетки, уменьшают поверхность катализатора. Чистоту исходных веществ в производстве азотной кислоты обеспечивают двумя путями - осуществлением дальнего забора воздуха и усовершенствованием систем очистки воздуха и аммиака.
В процессе окисления аммиака поверхность платиноидных сеток сильно разрыхляется, эластичные нити сеток становятся хрупкими. При этом поверхность сетки увеличивается примерно в 30 раз. Сначала это ведет к повышению каталитической активности катализатора, а затем к разрушению сеток. Срок жизни катализаторных сеток: для работы под атмосферным давлением – до 14 месяцев, под давлением 0,73 МПа – 8-9 месяцев.
Каталитическое окисление NH3 – гетерогенно-каталитический процесс, протекающий во внешнедиффузионной области и лимитируемый диффузией аммиака к поверхности катализатора. Скорость окисления очень высока. За десятитысячные доли секунды степень превращения аммиака достигает 97-98% при атмосферном давлении и 95-96% под давлением 0,88-0,98 МПа. Однако выход NO может быть различным в зависимости от выбранных технологических параметров – температуры, давления, линейной скорости газа, содержания аммиака в исходной смеси, числа сеток и других факторов.
Влияние температуры. На платине окисление аммиака начинается при 1950С, однако при этом образуется молекулярный азот. Заметное количество NO начинается появляться при 3000С. При повышении температуры выход NO увеличивается. Это свидетельствует о том, что энергия активации целевой реакции выше, чем энергия активации побочной реакции. Повышение температуры также приводит к увеличению скорости процесса. Но при большой температуре увеличиваются потери дорогостоящей платины. Повышение температуры с 780 до 8500С приводит к увеличению потерь катализатора вдвое. Величина оптимальной температуры зависит от используемого в процессе давления и составляет 780-9200С.
При выборе температуры также необходимо учитывать наличие примесей в исходной смеси. Температура должна быть тем выше, чем больше примесей.
Влияние давления. С ростом давления наблюдается снижение выхода NO. Вместе с тем использование высокого давления при окислении аммиака позволяет существенно уменьшить объем газовой смеси, а следовательно – габариты оборудования, т.е. увеличить производительность реактора при сохранении его объема. Поэтому наряду с технологическими схемами, работающими при атмосферном давлении, промышленное применение нашли агрегаты, работающие при давлении 0,41 – 0.73 МПа, в которых выход окиси азота увеличивают повышением температуры и времени контактирования (увеличение числа сеток).
Влияние концентрации аммиака. Согласно уравнению реакции, для полного окисления 1 моль аммиака необходимо 1,25 моль кислорода. Исходя из этого, максимально возможное содержание аммиака в аммиачно-воздушной смеси равно 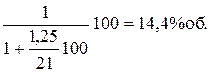
Однако при соотношении O2 : NH3 = 1,25 даже при атмосферном давлении выход NO не превышает 60-80%. Кроме того, при содержании в смеси 14.4% об. аммиака пришлось бы работать в области взрывоопасных концентраций. Нижний предел взрываемости амммиачно-воздушной смеси при атмосферном давлении составляет 13,8% об. NH3. При увеличении соотношения O2 : NH3 до 1,7, что соответствует содержанию аммиака в смеси 11,5%об. выход окиси азота увеличивается. Необходимость создания ~30% -ного избытка кислорода сверх стехиометрического связана с тем, что поверхность катализатора должна быть покрыта кислородом, в отсутствие кислорода аммиак уже при 500С начинает разлагаться на азот и водород. При дальнейшем увеличении соотношения реагентов выход целевого продукта меняется уже незначительно.
Влияние времени пребывания реагентов в реакторе. Для расчета времени контакта газов с катализатором пользуются уравнением
τ = Vсв./Vг,
где Vсв. – свободный объем катализатора, м3,
Vг – объемная скорость газа, м3/с.
Практикой показано, что оптимальное время пребывания газов в зоне катализа равно 10-4 – 10-5 с. Увеличение времени пребывания, т.е. уменьшение скорости газового потока, ведет к образованию элементарного азота. Увеличение скорости приводит к проскоку аммиака в поток нитрозного газа.
Реактор. Контактный аппарат представляет собой два усеченных конуса, соединенных между собой цилиндрической частью. В верхней части установлен фильтр с противовзрывным клапаном. Фильтр заключен в цилиндрическую обечайку, через которую подается аммиачно-воздушная смесь. Катализаторные сетки закреплены между фланцами цилиндрической части и нижнего корпуса и опираются на колосники. На решетке внизу аппарата расположен слой металлических колец, предназначенный для улавливания частиц платины и стабилизации теплового режима на катализаторных сетках.
Лекция № 9
Закономерности управления каталитическими процессами
Катализ – это явление изменения скорости химической реакции под воздействием малых количеств веществ – катализаторов, которые, участвуя в процессе, восстанавливают свой состав в конце каталитического цикла.
Катализ является наиболее эффективным и рациональным средством регулирования скорости химической реакции. Более 80% промышленных процессов являются каталитическими.
Катализатор может увеличивать скорость химической реакции (положительный катализ) или уменьшать ее (отрицательный катализ). В последнем случае катализатор называют ингибитором. Торможение (ингибирование) нежелательных процессов играет в химической технологии не менее важную роль, чем каталитическое ускорение реакции.
Ускоряющее действие катализаторов принципиально отличается от действия других факторов, интенсифицирующих процесс. Концентрация реагирующих веществ и давление увеличивают общее число столкновений молекул, температура, различные виды облучения увеличивают энергию сталкивающихся молекул. Катализатор снижает энергию активации процесса в результате изменения реакционного пути.
Пусть протекает реакция аА + вВ → сС + dD.
Чтобы вещества А и В образовали продукты С и D, они должны преодолеть некоторый энергетический барьер. На это затрачивается энергия активации Еа. Молекулы, обладающие этой избыточной энергией. Образуют неустойчивую группировку, называемую активированным комплексом АВ#. Скорость реакции непосредственно зависит от значения энергии активации; если она мала, то в единицу времени большее количество молекул преодолеют энергетический барьер, и скорость реакции будет высокой. Если энергия активации велика, то реакция идет медленно. Катализатор тем или иным способом изменяет реакционный путь. Например, он взаимодействует с молекулой А, образуя некоторый активированный комплекс АК#. Этот комплекс взаимодействует с молекулой вещества В, образуя новое неустойчивое соединение АВК#, которое, разрушаясь, дает продукты С и D и катализатор в первоначальном виде.
А + К → АК#
АК# + В → АВК#
АВК# → С + D + К
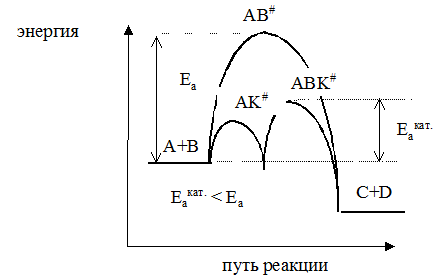
Таким образом, процесс разбивается на ряд стадий, каждая из которых требует преодоления меньшего энергетического барьера, чем в случае некаталитической реакции.
Мерой ускоряющего действия катализатора является величина относительной активности катализатора. Относительная активность рассчитывается как отношение константы скорости каталитической реакции к константе скорости некаталитической реакции.
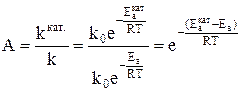
Видно, что даже незначительное уменьшение энергии активации может увеличить скорость реакции в десятки, сотни и более раз. В качестве примера можно привести реакцию окисления сернистого ангидрида, осуществляемую при температуре 693 К в присутствии катализатора V2O5.
Еа =420000 Дж/моль, Еакат. = 268000 дж/моль
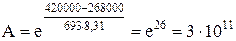
Скорость окисления SO2 в присутствии катализатора V2O5 возрастает в 3 . 1011 раз.
Даже, если реакция из-за своей малой скорости практически совсем не протекает. Использование подходящего катализатора приводит к полному и быстрому химическому превращению. Однако следует помнить, что:
1) Использование катализатора не может вызвать термодинамически невозможную реакцию. Если ∆G > 40 кДж/моль, реакция термодинамически невозможна, катализатор искать бесполезно. Если 0 < ∆G < 40 кДж/моль, и тем более, если ∆G < 0, подходящий катализатор искать можно и нужно.
2) Катализатор не может смещать положение равновесия в обратимых процессах, так как в равной степени ускоряет и прямую и обратную реакции, способствуя более быстрому достижению равновесия.
В зависимости от фазового состояния катализатора и реагентов различают катализ гомогенный и гетерогенный. Оценим преимущества и недостатки этих двух видов катализа.
| Характеристика | гомогенный катализ | гетерогенный катализ |
| 1. Вид системы | обычно жидкая среда с растворенным катализа-тором, редко газообраз-ные реагенты и газооб-разный катализатор | жидкая или газовая среда на твердом ката-лизаторе |
| 2.Изготовление катали-затора | Простое | сложное |
| 3. Воспроизводимость свойств катализатора | Высокая | сравнительно ниже |
| 4. Стоимость катали-затора | сравнительно низкая | высокая |
| 5.Селективность ката-лизатора | Высокая | сравнительно ниже |
| 6.Необходимость учета диффузионных факто-ров | отсутствует | необходимо создать условия для интенси-фикации массообмен-ных процессов |
| 7. Теплообмен с окру-жающей средой | легко организуемый | сложно организуемый |
| 8.Выделение катализа-тора из реакционной смеси | Сложное | простое |
Основным недостатком гомогенного катализа является сложность его выделения из реакционной среды. Часть катализатора, а иногда и весь катализатор теряется безвозвратно. Это увеличивает экономические затраты на производство, ухудшает качество продукта, увеличивает количество сточных вод и отходов.
Основными проблемами при использовании гетерогенного катализатора является необходимость решения вопросов, связанных с интенсификацией массо- и теплообменных процессов.
Методы управления гомогенно-каталитическими процессами мало чем отличаются от приемов интенсификации гомогенных некаталитических процессов, хотя участие катализатора в процессе вносит свою специфику.
Например, известно, что согласно закону действующих масс, скорость реакции должна возрастать пропорционально концентрации реагирующих веществ. Однако в гомогенно-каталитическом процессе А → С возможен случай, когда скорость реакции, увеличиваясь по мере увеличения концентрации реагента, достигает некоторой величины и перестает изменяться. Причиной этого является то, что общая скорость процесса

лимитируется стадией разрушения промежуточного комплекса катализатора с реагентом А. А + К → АК#
АК# → C + К
Скорость разрушения этого комплекса зависит от его концентрации, которая, в свою очередь, зависит от концентрации катализатора. Если концентрация катализатора мала, он весь связан в комплекс, и увеличение концентрации реагента А бесполезно.
Таким образом, следует помнить, что катализатор активно участвует в процессе; его концентрация может оказаться эффективным инструментом управления процессом.
Основные стадии и кинетические особенности гетерогенно-каталитических процессов

В общем случае процесс гетерогенного катализа складывается из следующих стадий:
1 – внешняя диффузия молекул реагентов из ядра потока к поверхности катализатора через пограничный слой δ;
2 – внутренняя диффузия молекул в порах катализатора;
3 – активированная адсорбция молекул на поверхности катализатора с образованием поверхностных непрочных химических соединений – активированных комплексов;
4 – перегруппировка атомов с образованием поверхностных комплексов «продукт-катализатор» (химическая реакция);
5 – десорбция молекул продуктов с поверхности;
6 – внутренняя диффузия молекул продуктов в порах катализатора;
7 – внешняя диффузия молекул продуктов от поверхности катализатора в ядро потока через пограничный слой.
Стадии 3, 4, 5 являются химическими, 1, 2, 6, 7 – массообменные (диффузионные).
В зависимости от того, какая из этих стадий является лимитирующей, различают три области протекания гетерогенно-каталитического процесса: внешнедиффузионную, внутридиффузионную и кинетическую.
Область протекания определяется экспериментально. 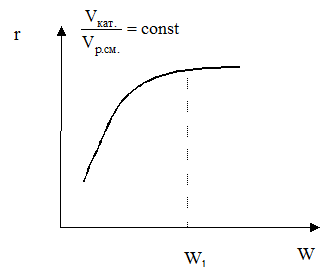
Если скорость процесса зависит от линейной скорости потока W при постоянном отношении объема катализатора и объема реакционной смеси, процесс протекает во внешнедиффузионной области, то есть лимитируется стадией внешней диффузии веществ через пограничный слой. Эта область является самой неблагоприятной для проведения процесса:
- работает только внешняя поверхность катализатора, которая значительно меньше внутренней;
- при экзотермических реакциях катализатор работает в жестком температурном режиме, так как выделяющееся тепло не успевает отводиться с поверхности катализатора;
- из-за жестких условий на поверхности катализатора селективность процесса низкая.
Для интенсификации процесса, протекающего во внешнедиффузионной области, используются такие инструменты управления, которые увеличивают скорость массопередачи в пограничном слое: повышение линейной скорости подачи реагентов, увеличение удельной поверхности контакта фаз (например, использование движущегося и «кипящего» слоя катализатора и др.).
В случае внутридиффузионной области лимитирующей является стадия движения молекул внутри пор. Это движение зависит от размера пор. В широких порах перенос вещества описывается законами молекулярной диффузии. В узких порах увеличивается вероятность ударов молекул о стенки канала, скорость движения зависит от диаметра этого канала, то есть описывается другими законами.
Во внутридиффузионной области химическая реакция и диффузия протекают одновременно, поэтому эту область можно назвать переходной между кинетической и внешнедиффузионной; на скорость процесса оказывают влияние как кинетические, так и диффузионные факторы.
В зависимости от соотношения скоростей внутренней диффузии и химической реакции внутренняя поверхность пор может использоваться полностью (если скорость химической реакции меньше) или частично (если протекает очень быстрая реакция). Внутридиффузионное торможение можно снять путем увеличения диаметра пор и уменьшения их длины.
Кинетическая область является наиболее благоприятной для ведения гетерогенно-каталитического процесса: работает вся поверхность катализатора, выделяющееся тепло легко отводится, достигается высокая селективность процесса. Скорость процесса равна скорости химической реакции, изменение которой подчиняется в этом случае законам хемосорбции.
Для ускорения процесса, протекающего в кинетической области, используют кинетические факторы.
Требования к гетерогенным катализаторам:
1) высокая каталитическая активность
2) высокая селективность
3) простота получения, обеспечивающая хорошую воспроизводимость свойств катализатора
4) высокая механическая прочность к сжатию, удару и истиранию
5) термическая стабильность (при превышении температуры на 50-100 катализатор не должен терять свои свойства)
6) большой срок службы и легкая регенерация
7) небольшая стоимость
Эффективность применения того или иного вещества в качестве катализатора определяется всей совокупностью его химических и физических свойств.
Химические свойства катализатора
Основными характеристиками катализатора, зависящими от его химического состава и свойств. Является каталитическая активвность и селективность.
Каталитическая активность измеряется скоростью превращения реагента, отнесенной к единице объема, поверхности или массы катализатора. Активность катализатора изменяется в процессе работы, и характер этого изменения зависит от природы катализатора и условий его работы. Кривая изменения активности во времени показана на рисунке.
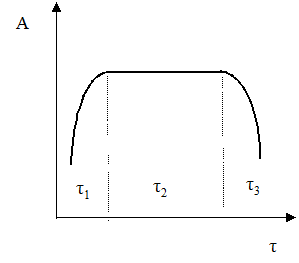
τ1 – период созревания катализатора; в это время под воздействием молекул реагента на поверхности происходят изменения, приводящие к повышению активности катализатора;
τ2 – период постоянной активности (срок службы катализатора);
τ3 – период дезактивации катализатора (потеря активности).
Причины дезактивации катализатора
| Вид дезактивации | Сущность процесса | Характеристика | Способы борьбы с потерей активности |
| 1.Старение катализатора | перестройка внутренней структуры катализатора, потеря его физических и механических свойств | необратимый, естественный, равномерный процесс | нет |
| 2.Утомление катализатора а) спекание б)зауглероживание в)минерализация г)отравление | потеря активности вследствие неправильной эксплуатации перегрев катализатора отложение на поверхности катализатора высококонденсированных ароматических структур (кокса) отложение на поверхности катализатора минеральных веществ блокировка активных центров на поверхности катализатора небольшими количествами веществ – каталитическими (контактными)ядами | неестественный, неравномерный процесс необратимый процесс обратимый процесс обратимый процесс отравление может быть обратимым и необратимым | 1)Соблюдение температурного режима; 2)организация правильного теплоотвода; 3)ввод структурообразующих добавок при приготовлении катализатора. 1)Профилактические меры: а)очистка сырья; б) введение в состав катализатора специальных добавок, препятствующих отложению; 2)Окислительная регенерация – обжиг катализатора в токе воздуха 1)Использование малочувствительных к каталитическим ядам катализаторов; 2)очистка сырья от каталитических ядов; 3)предкатализ - перевод токсичных примесей сырья в нетоксичные |
В качестве контактных ядов чаще всего выступают соединения элементов 5 и 6 групп (соединения мышьяка, сурьмы, кислорода, серы, селена, теллура), окись углерода СО, синильная кислота и ее соли, тяжелые металлы (медь, свинец, олово, висмут, ртуть, кобальт, никель, железо, марганец), галогены, хлорсодержащие углеводородов и др. Контактные яды часто обнаруживают некоторую специфичность. Так например, О2 является ядом для многих металлических катализаторов, а в случае платинового катализатора он даже увеличивает его активность. Никель, отравленный тиофеном, не гидрирует ароматические углеводороды, но сохраняет свою активность при гидрировании олефинов.
Действие контактных ядов зависит от их концентрации. Иногда для отравления катализатора достаточно очень малого количества яда. Например, присутствие 3 . 10-10 моль/л Н2S в сырье приводит к полной и необратимой дезактивации никелевого катализатора в процессе гидрирования. В других случаях отравление происходит при сравнительно больших концентрациях яда, а в малых количествах он повышает активность катализатора, промотирует его. (Промотирование – повышение активности катализатора путем введения различных добавок).
Отравление катализатора может быть обратимым и необратимым, прогрессирующим (постоянно возрастающим по мере накопления яда) и внезапным, быстрым. Отравление может быть селективным, т.е. включающим лишь часть функций катализатора, или полным.
Наиболее чувствительными к контактным ядам являются металлические катализаторы, особенно металлы 8 группы и металлы подгруппы меди. Из неметаллических катализаторов чувствительными к контактным ядам являются алюмосиликатные катализаторы.
Селективность катализатора – это способность катализатора ускорять только целевую реакцию. Селективность рассчитывается как отношение скорости образования целевого продукта к величине активности катализатора. В качестве примера использования селективного катализатора можно привести процесс селективного окисления аммиака до оксида азота (II) на платине (см. лекцию № 8).
Использование высокоселективных катализаторов позволяет уменьшить непроизводительные затраты сырья на побочные реакции, облегчает разделение реакционной смеси, то есть в целом улучшает экономику процесса.
Физические свойства катализатора
Физические свойства катализатора, такие как его поверхность, кристалличность, теплопроводность, механическая и термическая стабильность и др., оказывают очень сильное влияние на эффективность его работы, кинетику реакций, гидродинамику потоков.
Скорость гетерогенно-каталитической реакции, как скорость любого гетерогенного процесса, возрастает с увеличением поверхности контакта фаз, в данном случае – с увеличением поверхности катализатора, так как именно на его поверхности протекает реакция. Увеличить поверхность катализатора можно двумя способами:
- раздробить или размельчить катализатор
- увеличить его пористость.
При измельчении катализатора увеличивается его внешняя поверхность. Однако при уменьшении размера гранул катализатора возрастает гидравлическое сопротивление каталитического слоя, а, следовательно, и затраты на транспортировку реагентов через реактор. Кроме того, при высокой степени измельчения катализатора резко увеличивается его унос из реактора. Поэтому размер зерен катализатора должен иметь оптимальную величину. При неподвижном слое катализатора используют гранулы размером 2-5 мм, (редко до 20 мм); в «кипящем» слое катализатора размер гранул меньше 20 микрон. Поверхность гранул катализатора можно увеличить также, усложняя их форму. Однако это сопровождается, обычно, увеличением затрат на производство катализатора. Промышленные катализаторы имеют форму шаров, цилиндров, таблеток, колец Рашига и др.
Создание пористой структуры катализатора увеличивает внутреннюю поверхность, которая при развитой системе пор может составить более 75% от общей поверхности катализатора. Доступность внутренней поверхности определяется размером пор. Различают:
- макропоры (транспортные) - d = 1000-2000 А0, f = 0,5-2 м2/г
- средние (переходные) - d = 15 - 1000 А0, f = 400 м2/г
- микропоры – d соизмерим с размерами молекул, f > 400 м2/г.
Чем меньше размер пор, тем больше внутренняя поверхность. Однако размер пор должен быть таким, чтобы обеспечить наиболее благоприятные условия диффузии молекул реагентов внутри поры. Важное значение имеет также длина поры. Для каждого катализатора экспериментально подбирается оптимальная пористая структура, которая определяется соотношением скорости диффузии и скорости химической реакции.
На эффективность работы катализатора часто оказывает большое влияние его кристалличность. Так например, каталитическая активность γ-Al2O3 в реакциях дегидрирования и дегидратации в десятки раз больше, чем активность α- формы оксида алюминия, которая отличается от γ-формы температурным режимом приготовления.
Вполне очевидно, что срок службы катализатора и его активность в процессе работы очень сильно зависят от его механической прочности, теплопроводности, термостабильности. Поэтому в процессе приготовления катализатора особое внимание уделяют оптимизации этих физических свойств.
Классификация катализаторов
По механизму действия различают:
- ионные катализаторы, работающие по кислотно-основному механизму, то есть выступающие в роли кислоты или основания Льюиса;
- электронные катализаторы, работающие по окислительно-восстановительному механизму, то есть выступающие в роли окислителя или восстановителя;
- бифункциональные катализаторы.
В качестве ионных катализаторов используют протонные и апротонные кислоты на носителях (Н3РО4 на Al2O3, BF3 на Al2O3, гетерополикислоты, ионообменные смолы), природные и синтетические алюмосиликаты (Al2O3)m(SiO2)n(H2O)p, нейтральные и кислые соли Са3(РО4)2, СаНРО4, МgНРО4, оксиды некоторых металлов Al2O3, W2O3 и др. Кислотно-основной катализ обычно реализуется в реакциях, сопровождающихся гетеролитическим разрывом ковалентной связи. Это процессы гидратации, аминирования, алкилирования, изомеризации и т.д.
В качестве электронных катализаторов используют металлы (платину, серебро, родий, палладий, никель) и некоторые оксиды металлов (MgO, ZnO, Fe2O3, Cr2O3, WO3, MoO3, V2O5). Окислительно-восстановительный катализ наблюдается при гомолитическом разрыве связей в процессах гидрирования, дегидрирования, синтеза аммиака, окиси этилена, серной кислоты и др.
К бифункциональным катализаторам, используемым в процессах риформинга, гидрокрекинга и др., относятся Pt на Al2O3, MoO3 на Al2O3, смесь ZnO, Al2O3 и MgO.
По составу различают следующие группы катализаторов:
- смешанные
- модифицированные
- катализаторы на носителях (трегерные).
Смешанные катализаторы представляют собой смесь, состоящую из нескольких компонентов. Каждая отдельная часть смеси выполняет свою функцию. Очень часто при смешении наблюдается явление синергизма: активность смешанного катализатора выше, чем суммарная активность отдельных его компонентов.
Модификация – это способ увеличения активности и селективности катализатора за счет введения в его состав специальных добавок - модификаторов (или промоторов). Количество вводимых добавок очень небольшое, характер действия зависит от природы добавки. Различают:
1) модификаторы, увеличивающие активность катализатора,
2)структурообразующие добавки, изменяющие величину и характер поверхности катализатора,
3)упрочняющие добавки, увеличивающие механическую прочность и термостабильность катализатора,
4)блокирующие добавки, нейтрализующие каталитические яды.
Деление модификаторов на перечисленные группы весьма условно; часто (и это желательно) добавка выполняет несколько функций.
Катализаторы на носителях представляют собой каталитически активное вещество, нанесенное на инертный носитель с сильно развитой поверхностью. Функции носителя сводятся к увеличению поверхности катализатора, предохранению от спекания и разрушения, улучшению теплопроводности, иногда носитель играет роль активатора. В качестве носителей используют пемзу, кизельгур, асбест, синтетические материалы. Очень часто на носитель наносят дорогостоящие металлы: платину, серебро, палладий и др.
По методу приготовления промышленные катализаторы делят на следующие группы:
- природные (силикаты и алюмосиликаты),
- контактные массы, получаемые механическим смешиванием компонентов,
- осажденные, получаемые осаждением катализатора в виде геля из водного раствора соли под воздействием различных осадителей с последующей сушкой его и прокаливанием,
- катализаторы на носителях, получаемые путем пропитки носителя,
- плавленые в виде проволочных сеток, спиралей и т.д.,
- скелетные (металлические), в частности никель Ренея, который получают выщелачиванием никель-алюминиевого сплава избытком горячей щелочи.
Более 80% катализаторов получают методом осаждения.
Особую группу катализаторов составляют ферменты, которые представляют собой гомогенные биологические катализаторы белковой природы, очень сложные по составу. Основными достоинствами этих катализаторов является их высокая активность (реакции, катализируемые ферментами, протекают при комнатной температуре) и стереоспецифичность, то есть способность реагировать только с одним фрагментом молекулы в реакционной смеси.
Лекция №10
Дата добавления: 2016-01-20; просмотров: 4221;