Общая характеристика и классификация каталитических реакций
Скорость многих химических реакций зависит от присутствия в реакционной смеси веществ (специально вводимых или присутствующих в виде примесей), которые не входят в стехиометрическое уравнение химической реакции и после окончания реакции остаются неизменными как по химическому составу, так и количественно. Эти вещества, изменяющие скорость реакции, но стехиометрически не являющиеся участниками реакции, получили название катализаторы(Берцелиус, 1836), а само явление изменения скорости в присутствии катализатора называется катализом. Каталитические реакции очень широко распространены как в химической технологии, так и в биологических процессах (ферментативный катализ). Многие реакции, которые на первый взгляд не являются каталитическими, в действительности ускоряются очень малыми количествами примесей (например, следовыми количествами воды или других веществ), всегда присутствующих в реакционной смеси, а в некоторых случаях катализатором может быть даже сам реакционный сосуд.
Влияние катализатора на скорость реакции обусловлено тем, что он входит в состав активного комплекса и изменяет энергию активации химической реакции.
При распаде активного комплекса происходит регенерация катализатора, т.е. теоретически катализатор не расходуется и остается химически неизменным. Поэтому в принципе одной малой порции катализатора достаточно для проведения реакции с любым количеством субстрата (реагирующего с катализатором вещества). Конечно, в реальных процессах катализатор расходуется из-за протекания побочных процессов, отравления, технологических потерь, изменения состояния поверхности твердых катализаторов и т.п.
Одним из важнейших свойств катализаторов является их селективность(избирательность) – каждый катализатор влияет на скорость одной реакции или какой-либо группы реакций. Различные катализаторы могут образовывать с одними и теми же исходными веществами как однотипные, так и различные по составу и структуре активные комплексы. Это может привести к изменению направления процесса и к образованию из одних и тех же исходных веществ различных продуктов.
Сказанное выше не означает, что катализатор может вызвать термодинамически невозможный в данных условиях процесс или повлиять на положение равновесия в обратимых реакциях. Суть в том, что если в данных условиях термодинамически возможно протекание нескольких параллельных реакций, то катализатор вследствие свойства селективности ускоряет одну из этих реакций. Положение равновесия для нее достигается значительно быстрее, чем для других реакций, поэтому основными продуктами будут вещества, образующиеся в этой реакции, а продукты других реакций будут присутствовать в качестве примесей к основным.
В некоторых случаях сами продукты реакции могут быть катализаторами и реакция самоускоряется с течением времени. Такие реакции называются автокаталитическими.
При протекании реакции катализатор и реагирующие вещества могут находиться в одной фазе (жидкой или газообразной), и такие реакции являются гомогенными каталитическими. Наиболее часто гомогенный катализ наблюдается в растворах. В случае гетерогенного катализа катализатор и реагирующие вещества находятся в разных фазах – в большинстве случаев катализатор твердый, а реагирующие вещества находятся в газообразной фазе или растворе.
24.2. Гомогенные каталитические реакции
Поскольку катализатор не входит в стехиометрическое уравнение химической реакции, но оказывает влияние на ее скорость, логично предположить, что катализатор вначале образует с реагирующими веществами некоторые неустойчивые промежуточные соединения, которые при дальнейших превращениях дают продукты реакции с регенерацией катализатора. Иногда промежуточные продукты представляют собой обычные, но малоустойчивые химические соединения, которые можно выделить и изучить отдельно. Однако в большинстве случаев чрезвычайно малая устойчивость промежуточных продуктов не позволяет их выделить и об образовании промежуточного соединения можно судить иногда лишь косвенно по изменению какого-нибудь свойства раствора. Например, при реакции метафосфорной кислоты с персульфатом калия, катализатором которой является йодоводородная кислота, наблюдается появление желтой окраски раствора, которая исчезает к моменту окончания реакции:
H3PO3 + K2S2O8 + H2O 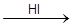 H3PO4 + K2SO4 + H2SO4
H3PO4 + K2SO4 + H2SO4
Промежуточным продуктом здесь является йод и реакция протекает в две стадии:
1) K2S2O8 + 2HI ® I2 + H2SO4 + K2SO4
2) H3PO3 + I2 + H2O ® H3PO4 + 2HI
Окисление тиосульфат-иона перекисью водорода катализируется молибденовой кислотой:
S2 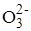 + 4H2O2
+ 4H2O2 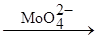 2S
2S 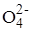 + 2H+ + 3H2O
+ 2H+ + 3H2O
Предполагается, что промежуточным продуктом является пермолибдат-ион и реакция протекает по схеме:
1) 4Mo + 4H2O2 ® 4Mo 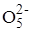 + 4H2O
+ 4H2O
2) S2 + 4Mo + H2O ® 2S 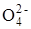 + 2H+ + 4Mo
+ 2H+ + 4Mo
Исходя из представлений теории активированного комплекса, предполагается следующая схема каталитического действия.
Некаталитическая реакция между веществами А и В протекает с образованием активированного комплекса АВ#:
А + В ® АВ# ® C + D
В присутствии же катализатора K процесс идет в несколько стадий:
1) Обратимая реакция образования промежуточного продукта из катализатора и какого-либо реагирующего вещества:
А + K 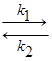 АK
АK
2) Промежуточный продукт образует активированный комплекс:
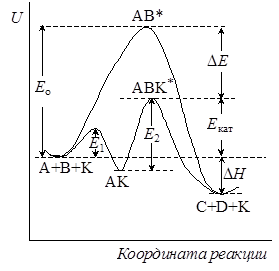 Рис. 24.1. Профили пути некаталитической и каталитической реакций.
Рис. 24.1. Профили пути некаталитической и каталитической реакций.
|
АK + В 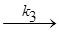 АВK#
АВK#
3) Активированный комплекс распадается на продукты реакции и катализатор:
АВK# 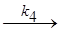 С + D + K
С + D + K
В принципе можно построить поверхность потенциальной энергии и представить профили путей некаталитической и каталитической реакций. Примерная их схема изображена на рисунке 24.1.
Как видно из рисунка, при катализе в данном случае предполагается, что образование промежуточного продукта является экзотермическим процессом, а далее потенциальная энергия системы возрастает в связи с образованием активированного комплекса АВК#. Из рисунка видно также снижение энергии активации каталитической реакции по сравнению с некаталитической DE = Eо – Eкат . Если считать, что предэкспоненциальные множители в уравнении Аррениуса для обеих реакций одинаковы, то отношение констант скоростей каталитической и некаталитической реакций равно
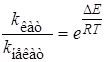 . (24.1)
. (24.1)
Так как величина DE стоит в показателе степени, то даже небольшое снижение энергии активации приводит к значительному возрастанию скорости реакции. Например, для реакции разложения ацетальдегида
СН3СНО ® СН4 + СО,
протекающей при 800К в газовой фазе, энергия активации равна примерно 190 кДж/моль. Реакция катализируется парами йода, при этом энергия активации снижается до 135 кДж/моль, т.е. DE = 55 кДж/моль (» 28%). Применяя формулу (24.1), получим
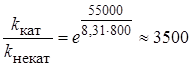 ,
,
т.е. в присутствии катализатора реакция идет в 3500 раз быстрее. Кинетика гомогенной каталитической реакции, протекающей по представленной выше схеме, зависит от свойств промежуточного продукта АК. Для вывода кинетического уравнения реакции представим скорость образования конечного продукта С как скорость мономолекулярного распада активированного комплекса:
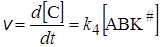 . (24.2)
. (24.2)
Используя принцип стационарных концентраций, определим концентрацию активированного комплекса из соотношения
k3[АK][В] = k4[АВK#]. (24.3)
Принцип стационарности применим также к промежуточному продукту АK:
k1[А][K] – k2[АK] – k3[АK][В] = 0. (24.4)
Общую концентрацию катализатора [Kо] можно представить как сумму текущей концентрации свободного катализатора [K] и концентрации промежуточного продукта [АK]:
[Kо] = [K] + [АK] = const. (24.5)
Тогда уравнение (24.4) можно записать в виде
k1[А]([Kо] – [АK]) – k2[АK] – k3[АK][В] = 0, (24.6)
откуда
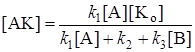 . (24.7)
. (24.7)
Определив по уравнениям (24.3) и (24.7) концентрацию [АВК#] и подставив ее в уравнение (24.2), получим уравнение для скорости реакции:
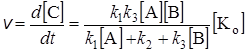 . (24.8)
. (24.8)
Это уравнение лежит в основе описания кинетики гомогенных каталитических реакций. Оно устанавливает пропорциональность между скоростью реакции и концентрацией катализатора, что подтверждается опытом.
При анализе этого уравнения можно рассмотреть некоторые предельные случаи. Промежуточный продукт может быть неустойчивым, и константа скорости k2 обратного разложения АK может оказаться очень большой, т.е. промежуточный продукт в основном разлагается на исходные вещества, его концентрация мала, поэтому и скорость его превращения в активированный комплекс и далее в конечные продукты очень мала (k2 >> k3[В]). В этом случае концентрация промежуточного продукта будет близка к равновесной, и в знаменателе уравнения (24.8) можно пренебречь величинами k1[А] и k3[В] по сравнению с k2. Тогда
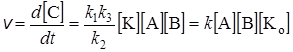 , (24.9)
, (24.9)
т.е. в этом случае реакция проходит по второму порядку относительно исходных веществ со скоростью, пропорциональной концентрации катализатора. Промежуточный продукт AK с такими свойствами называется промежуточным продуктом Аррениуса. Это название связано с тем, что активная форма молекулы, существование которой предполагалось при выводе зависимости скорости реакции от температуры (см. раздел 22.7), относится к соединениям этого типа.
В другом крайнем случае возможно образование промежуточного продукта, который будет по мере образования немедленно реагировать дальше в сторону образования активированного комплекса и продуктов реакции (k1[A]<<k3[В]и k2<<k3[В]). В этом случае скорость реакции
v = k1[A][Kо] (24.10)
и не зависит от концентрации второго исходного вещества, так как при любых, даже самых малых, концентрациях его достаточно для немедлен 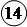 ного потребления АK. Промежуточные соединения такого типа называются веществами Вант-Гоффа, так как со времени Вант-Гоффа образованием таких веществ объясняют понижение общего порядка реакции по сравнению с возможными предположениями. Можно сказать, что в данном случае реакция имеет нулевой порядок по веществу B. Концентрация вещества Вант-Гоффа, в отличие от вещества Аррениуса, далека от равновесной.
ного потребления АK. Промежуточные соединения такого типа называются веществами Вант-Гоффа, так как со времени Вант-Гоффа образованием таких веществ объясняют понижение общего порядка реакции по сравнению с возможными предположениями. Можно сказать, что в данном случае реакция имеет нулевой порядок по веществу B. Концентрация вещества Вант-Гоффа, в отличие от вещества Аррениуса, далека от равновесной.
Предложены также другие схемы хода гомогенных каталитических реакций, но во всех случаях скорость реакции зависит от концентрации промежуточного продукта, а значит, и от концентрации катализатора. Если, например, промежуточный продукт Р получается из исходного вещества А и катализатора K по схеме
n + K L Р,
то константа равновесия этой реакции
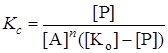 , (24.11)
, (24.11)
а концентрация промежуточного продукта
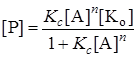 . (24.12)
. (24.12)
Общая скорость реакции зависит от скорости реакции разложения промежуточного продукта
Р 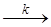 продукты реакции + K
продукты реакции + K
Итак, скорость реакции
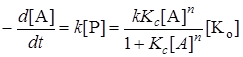 . (24.13)
. (24.13)
Если равновесие смещено в сторону образования промежуточного продукта, то есть величина Kс[A]n >> 1, то скорость реакции
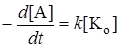 . (24.14)
. (24.14)
Если же равновесомая смещенная в сторону исходных веществ (Kс[A]n << 1), то
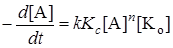 . (24.15)
. (24.15)
Константа равновесия Kс зависит от температуры, поэтому при изменении температуры возможно изменение порядка реакции по реагирующим веществам. Такое изменение обнаружено, например, для реакции разложения пероксиду водорода, которое катализируется ионами 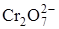 . В интервале температур от 0 до 56 оС порядок этой реакции по Н2О2 изменяется от нулевого до второго.
. В интервале температур от 0 до 56 оС порядок этой реакции по Н2О2 изменяется от нулевого до второго.
24.3. Кислотно-основный катализ
В многих случаях реакции в растворах катализируются ионами водорода и гидроксила. Еще в 1884 г. Оствальд установил, что сила каталитического действия кислот пропорциональна их электрической проводимости. Это правило подтвердил Аррениус, который обнаружил также два новых эффекта каталитического действия.
Добавление нейтральной соли, которая не имеет общего катиона с кислотой-катализатором, повышает ее каталитическое действие – первичный солевой эффект. Например, скорость инверсии тростникового сахара в водном растворе уксусной кислоты (катализатор) возрастает приблизительно на 30% при добавке 10 мол. % NaCl.
Если к раствору кислоты прибавлять ее соль, то в соответствии с общей теорией электролитической диссоциации концентрация ионов водорода должна уменьшаться, а каталитическое действие кислоты ослабевать. На самом деле же в некоторых реакциях каталитический эффект не только не уменьшается, а даже усиливается (например, при этерификации трихлоруксусной кислоты). Это явление получило название вторичного солевого эффекта. Оно свидетельствует о том, что каталитическое действие проявляют недиссоциированные молекулы кислоты и ее анионы.
В зависимости от природы катализаторов различают три типа кислотного или основного катализа:
1) специфический кислотный катализ, в котором катализатором являются ионы Н3О+, и специфический основный катализ ионами ОН–;
2) общий кислотный катализ, в котором катализаторами выступают любые доноры протонов, кроме ионов Н3О+, то есть обобщенные кислоты Бренстеда, и общий основной катализ под действием любых акцепторов протонов, кроме ионов ОН–, то есть под действием обобщенных оснований Бренстеда;
3) электрофильный катализ, где катализаторами выступают кислоты Льюиса (акцепторы свободной пары электронов), и нуклеофильный катализ, в котором катализаторами выступают основания Льюиса (доноры свободной пары электронов).
Примером специфического кислотного катализа может быть реакция гидролиза сложных эфиров в сильнокислых растворах. Скорость этой реакции зависит от концентрации ионов Н3О+ и эфира:
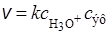 .
.
Предложена следующая схема этой реакции, которая состоит из нескольких стадий:
 O O H
O O H
 R – C – OR' + H3O+ L R– C –
R – C – OR' + H3O+ L R– C –  + H2O
+ H2O
 R'
R'


 О H О– H
О H О– H


 H2O + R – C –
H2O + R – C –  R – C –
R – C –

 R' О+ R'
R' О+ R'
Н Н
 O
O
R – C – H2 + R'OH
 O O
O O
R – C – H2 + H2O  R – C – OH + H3O+
R – C – OH + H3O+
Как видно из этой схемы, сначала протон быстро переходит от иона Н3О+ к спиртовому атому кислорода, потом в результате взаимодействия с молекулой воды получается промежуточное соединение ионного типа, которое распадается на молекулу спирта и положительно заряженный ион RCOO 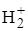 . Последний при реакции с молекулой воды образует кислоту и ион Н3О+, который снова может вступать в реакцию.
. Последний при реакции с молекулой воды образует кислоту и ион Н3О+, который снова может вступать в реакцию.
Схема специфического гидролиза эфиров в щелочной среде может быть представлена таким образом:
 O O O
O O O



 R – C – O – R' ® R – Cd– O
R – C – O – R' ® R – Cd– O 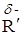 ® R – C – OH + R'OH + OH–
® R – C – OH + R'OH + OH–

 HO– H–O–H HOd– H Od– H
HO– H–O–H HOd– H Od– H
В этой реакции ион гидроксила взаимодействует с карбонильным атомом углерода, а молекула воды – с атомом кислорода спиртовой группы – реакция переноса протона (d– – частичный заряд). Промежуточное соединение можно считать активированным комплексом.
Общий кислотный катализ, как и специфический, также связан с взаимодействием протона с молекулой субстрата, а донором протона может быть любая кислота Бренстеда НА. В многих случаях при этом наиболее медленной стадией является не распад, а образование катиона субстрата SH+. Для общего основного катализа также характерной является, в отличие от специфического катализа, медленная скорость образования промежуточного активированного комплекса S-.
Надо заметить, что установления истинного механизма реакций представляет собой довольно сложную задачу, поэтому различные схемы, которые предлагаются для объяснения кинетики реакций, можно рассматривать как наиболее вероятные, но они могут измениться при дальнейших, более детальных исследованиях.
24.4. Общая характеристика гетерогенных каталитических
процессов
В гетерогенных процессах реагирующие компоненты и катализатор находятся в разных фазах и реакция протекает в пограничном слое – на границе раздела фаз. На практике чаще всего катализатор находится в твердой фазе, а реагирующие вещества – в жидкой или газообразной фазе. Поэтому для гетерогенных каталитических реакций особое значение имеет транспорт реагирующих веществ из объема жидкой или газообразной фазы к поверхности катализатора и их адсорбция. Весь процесс можно разделить на несколько этапов: 1) транспорт реагирующих веществ к поверхности катализатора (диффузия); 2) адсорбция реагирующего вещества на поверхности катализатора; 3) реакция на поверхности катализатора; 4) десорбция продуктов с освобождением поверхности катализатора; 5) отвод продуктов в газовую или жидкую фазу. В зависимости от условий проведения процесса и его особенностей наиболее медленной может быть любая из этих стадий, а, следовательно, и скорость каталитического процесса может лимитироваться одной из них. Поэтому в тех случаях, когда определяется активность катализатора, процесс нужно проводить таким образом, чтобы скорость его определялась третьей стадией.
Для объяснения каталитического действия была выдвинута идея (Менделеев, Сабатье, Зелинский) об образовании промежуточных продуктов. Согласно этой идее, катализатор образует с одним из реагирующих веществ промежуточное соединение, активируя данный реагент и облегчая реакцию. Типичными промежуточными соединениями являются сорбционные соединения типа 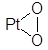 , Ni–H, Pd–H и т.д.
, Ni–H, Pd–H и т.д.
Отсюда следует и один из основных принципов гетерогенного катализа: катализатор обладает сродством к одному или нескольким реагентам. Сродство катализатора к реагирующим веществам и обеспечивает образование промежуточного соединения, которое во многих случаях является неустойчивым и распадается в дальнейших превращениях на конечные продукты с регенерацией катализатора.
Как и в случае гомогенного катализа, катализатор в гетерогенных реакциях обладает свойством селективности (избирательности). Для одних и тех же веществ различные катализаторы ускоряют одну из термодинамически возможных в данных условиях реакцию, что приводит к образованию в качестве основного вещества различных конечных продуктов (другие вещества могут присутствовать в качестве примесей). Например, из смеси оксида углерода с водородом (водяной газ) в зависимости от катализатора и условий проведения опыта могут образовываться различные продукты. Если реакция проводится над металлическим никелем при 510 – 530 К, то получается метан:
СО + 3Н2 ® СН4 + H2O
На меди под повышенным давлением образуется метиловый спирт:
СО + 2Н2 ® СН3ОН
а на металлическом кобальте получается смесь высших олефинов и парафинов.
Различные продукты можно получить из этилового спирта при его каталитических превращениях в зависимости от выбора катализатора и условий протекания (температуры, давления):
СН2=СНСН=СН2 + Н2 +2Н2О
ZnO,
½ Cr2O3
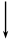

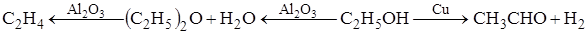
 активированная Cu ½Na
активированная Cu ½Na
¯
CH3COOC2H5 + 2H2 C4H9OH + H2O
½активированнаяCu
¯
CH3COCH3 + 3H2 + CO
Причины избирательного действия катализаторов в каждом случае могут быть разными. По теории промежуточных продуктов это действие связано с образованием промежуточных соединений разной химической природы на разных катализаторах.
Часто для придания катализатору большей избирательности, повышения его термической стойкости и механической прочности, а также повышения активности катализаторы применяют не в виде чистых веществ, а в виде сложных многокомпонентных систем. Смешанные катализаторы обычно представляют собой смесь двух или нескольких окислов, например, Al2O3 + ThO2; Al2O3 + Cr2O3; CoO + MgO. Активность таких катализаторов является обычно функцией состава и при определенном содержании компонентов достигается максимальная активность.
Активность катализатора может быть увеличена при добавлении к нему вещества, которое само по себе не обладает каталитическими свойствами. Такие вещества получили название промоторов, а само явление – промотирования. Различают два типа промотирующего действия – структурообразующее промотирование и модифицирование. Структурообразующие промоторы стабилизируют активную фазу катализатора по отношению к температуре или каким-либо другим факторам. Видимо, они увеличивают срок жизни микрокристаллической фазы катализатора, неустойчивой вследствие термодинамически самопроизвольного термического укрупнения кристалликов катализатора. Примером может быть промотирование оксидом алюминия железного катализатора синтеза аммиака. Добавление небольших количеств Al2O3 вдвое увеличивает первоначальную активность катализатора и поддерживает его стабильность в течение длительного времени. Модифицирующие промоторы изменяют строение и химический состав активной фазы. Их действие сводится, вероятно, к синтезу на поверхности катализатора активных центров новой химической природы, при этом иногда наблюдается и изменение селективности катализатора.
Присутствие в реакционной системе некоторых веществ, часто в чрезвычайно малых количествах, может значительно снижать или полностью подавлять активность катализатора. Такие вещества получили название каталитических ядов, а само явление – отравление катализаторов. Типичными каталитическими ядами являются соединения серы, синильная кислота и некоторые ее производные, оксид углерода, свободные галогены, ртуть и некоторые ее соли, соединения фосфора, мышьяка, свинца и др. и даже пары воды. Отравление катализатора в основном происходит в результате адсорбции яда на поверхности и блокировки активных центров катализатора. Так как адсорбция может быть обратимой и необратимой, то и отравление катализатора может быть обратимым и необратимым. При обратимом отравлении активность катализатора восстанавливается после удаления яда, а при необратимом восстановить активность катализатора не удается. Наибольшее отравляющее действие оказывают первые порции яда, которые резко, на 70–80%, снижают активность катализатора, после чего токсичность яда снижается. Изотермы отравления обычно удовлетворяют экспоненциальной зависимости:
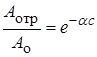 , (24.16)
, (24.16)
где Аотр – активность отравленного катализатора; Ао – начальная активность; с – количество яда, адсорбированного катализатором; a – коэффициент отравления, зависящий от природы яда, а также природы и свойств катализатора и некоторых параметров процесса. При малых количествах яда справедливо линейное приближение:
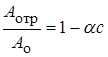 . (24.17)
. (24.17)
Исследования процессов отравления сыграли значительную роль в развитии теории катализа. Практическим же следствием явилась разработка требований к чистоте исходных веществ и аппаратуре для проведения каталитических реакций.
24.5. Активация в гетерогенных каталитических реакциях
В большинстве случаев для константы скорости гетерогенной каталитической реакции выполняется уравнение Аррениуса:
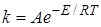 . (24.18)
. (24.18)
Однако, даже если каталитическая и некаталитическая реакции протекают одинаково, предэкспоненциальные множители обеих реакций могут быть разными, а энергии активации тоже не одинаковы. Поэтому отношение констант скоростей каталитической и некаталитической реакций равно:
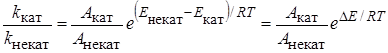 , (24.19)
, (24.19)
где Eкат – определенная экспериментально из температурной зависимости константы скорости энергия активации, которую называют кажущейся энергией активации.
Сопоставим кинетику некоторой бимолекулярной реакции между веществами А и В, протекающую без катализатора и в его присутствии. Без катализатора ход процесса представим схемой:
А + В ® АВ# ® Продукты
Каталитический процесс можно представить следующим образом:
1) Адсорбция исходных веществ на поверхности катализатора:
| |
Этот процесс, как правило, активированный (с энергией активации Еадс) и экзотермический, т.е. состояние АВK обладает меньшей потенциальной энергией, чем состояние (А + В + K).
2) Переход адсорбционного комплекса в активированное состояние:
АВК ® АВК#
Этот процесс требует затраты определенной энергии Еист, называемой истинной энергией активации гетерогенной каталитической реакции.
3) Реакция адсорбированного активированного комплекса с образованием конечных продуктов, адсорбированных на катализаторе:
АВК# ® (продукты)K
4) Десорбция продуктов реакции и регенерация катализатора:
(продукты)K ® Продукты + K
Этот процесс также активированный с энергией активации Едес, но эндотермичный.
 Рис. 24.2. Схема профиля пути гетерогенной каталитической реакции
Рис. 24.2. Схема профиля пути гетерогенной каталитической реакции
|
На рисунке 24.2 схематически изображен профиль пути гетерогенной каталитической реакции, протекающей по описанной схеме.
Как видно из рисунка, величина DЕ представляет собой изменение энтальпии при адсорбции активированного комплекса на катализаторе. Но такое истолкование справедливо лишь в предположении, что активированные комплексы в каталитической и некаталитической реакциях аналогичны. В общем случае эта величина может включать также изменения энтальпии, обусловленные энергетическими и конфигурационными перестройками внутри активного комплекса.
24.3. Кинетика гетерогенных каталитических реакций
в статических условиях
Рассмотрим гетерогенные каталитические реакции с газообразными веществами, реагирующими на твердом катализаторе. В качестве примера реакций такого типа могут служить реакции разложения некоторых веществ, например, распад аммиака на платине, метана на угле и т.п.
Как было сказано, в типичном гетерогенном процессе реагируют только те вещества, которые адсорбированы на поверхности катализатора. Скорость v гетерогенной химической реакции определяется как количество вещества, реагирующего в единицу времени на единице площади поверхности катализатора:
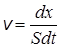 , (24.20)
, (24.20)
где dx – количество вещества, прореагировавшего к моменту времени t, моль; S – общая площадь поверхности катализатора.
Скорость химической реакции, согласно основному постулату химической кинетики, прямо пропорциональна поверхностной концентрации веществ. Последняя величина прямо пропорциональна степени заполнения поверхности q (см. гл. 12), поэтому можно записать, что
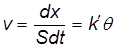 (24.5)
(24.5)
Так как поверхность данного катализатора постоянна, то ее величину можно ввести в константу скорости процесса k’, т.е. принять
k'S = k.
Поэтому уравнение (24.21) перепишем в форме:
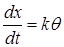 . (24.22)
. (24.22)
При условии установления адсорбционного равновесия величину поверхности, занятую реагирующим веществом на единице поверхности катализатора, можно найти из изотермы адсорбции Ленгмюра (см. раздел 12.5):
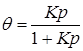 . (24.23)
. (24.23)
Из последнего уравнения следует, что если адсорбция мала, то q << 1, и, следовательно, K <<1, поэтому величиной Kp в знаменателе уравнения можно пренебречь и
q = Kp.
Тогда скорость реакции
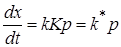 . (24.24)
. (24.24)
В этом случае кинетическое уравнение гетерогенной каталитической реакции соответствует уравнению реакции первого порядка.
При значительной адсорбции величина Kp >> 1 и q » 1. Тогда скорость реакции
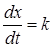 (24.25)
(24.25)
не зависит от концентрации реагирующего вещества, т. е. кажущийся порядок реакции является нулевым.
Следует заметить, что кажущийся порядок гетерогенных реакций может изменяться для одной и той же реакции от нулевого через дробный переменный порядок до первого при переходе от больших давлений к более низким.
Константа адсорбционного равновесия K зависит от природы вещества, которое адсорбируется, природы катализатора и характера его поверхности. Если реагирующее вещество образует с катализатором прочный адсорбционный комплекс, то катализирующее действие будет слабым, так как в таком случае затрудняется переход комплекса в активированное состояние и дальнейшие его преобразование в продукты реакции. Незначительное каталитическое действие будет наблюдаться также при образовании слабых связей между реагирующим веществом и катализатором и малой адсорбции, поскольку энергия активации будет уменьшаться в незначительной степени. Оптимальным будет катализатор, который образует с реагирующим веществом адсорбционный комплекс с некоторой промежуточной прочностью связей.
| <== предыдущая лекция | | | следующая лекция ==> |
| Волновая поверхность | | | Загальні відомості про видобування сировини |
Дата добавления: 2015-12-16; просмотров: 4300;