Последствия транспортных или ферментативных нарушений
Значение генетического дефекта для метаболизма клетки и состояния организма зависит от роли мутантного белка в обмене веществ и тяжести дефекта. Как уже упоминалось, большинство врожденных «ошибок» — результат дефекта внутриклеточных ферментов или процессов мембранного транспорта. Поскольку подобного рода мутации неоднократно обсуждаются далее, целесообразно очертить возможные последствия нарушения транспортных или ферментативных процессов в обобщенном виде. Примером может служить схематическая последовательность реакций, представленная на рис. 305-1. Литерами А, В, С, D, F и G обозначены субстраты или продукты ряда ферментативных реакций, а та, ЕАВ, ЕВС
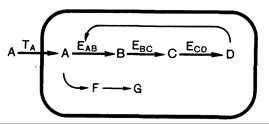
Рис.305-1. Схематическое изображение метаболического пути, включающего в себя систему транспорта (Тд), ферменты, альтернативные реакции и регуляцию по принципу обратной связи (по Rosenberg).
А, В, С, D — субстраты и продукты основного пути; F, G — продукты малого пути, та — транспортная система для А; ЕАВ ЕВС и ecd— ферменты, катализирующие превращение А в В,.В в С и С в D. и ecd — специфические транспортные системы или ферменты, катализирующие отдельные реакции этого ряда. Главный путь включает в себя превращение А в D через интермедиаты В и С; F и G — это продукты альтернативного пути метаболизма. Стрелка от D к ЕAB отражает отрицательную обратную связь между конечным продуктом последовательности реакций и первым ферментом этого пути. В дальнейшем будут по возможности приводиться примеры конкретных врожденных «ошибок», иллюстрирующие специфические последствия нарушения транспорта или ферментативных реакций.
Недостаточность предшественника. Если дефект локализуется на уровне та, т. е. в рецепторной или транспортной системе, переносящей А в клетку, то внутриклеточная концентрация А может оказаться слишком малой, чтобы насытить ЕAB субстратом. Это могло бы замедлять всю последовательность реакций и приводить к недостаточному образованию В, С и D. При болезни Хартнупа (см. гл. 308) нарушается транспорт триптофана в кишечнике. Эта транспортная аномалия имеет серьезные химические и клинические последствия, поскольку триптофан внутри клетки превращается в никотинамид. У больных с этой патологией могут наступить мозжечковая атаксия и развиться временная или постоянная деменция из-за недостаточности никотинамида, если они дополнительно не получают ниацин с пищей. Подобно этому, у больных с врожденным нарушением всасывания витамина B12 в кишечнике развивается мегалобластическая анемия, если им не вводить витамин парентерально. Недостаточность предшественника или субстрата может иметь место и в случае нарушения на уровне циркулирующего белка, переносящего вещество А в крови и доставляющего его к поверхности клетки.
Накопление предшественника. Рассмотрим эффект сниженной активности одного из внутриклеточных ферментов (еаb, ЕBC или ЕCD). Этот дефект мог бы приводить к накоплению внутри и вне клетки ближайших или отдаленных предшественников реакции. Если нарушен ЕAB, накапливается только А. Эта ситуация иллюстрируется резким увеличением количества лизосомного глюкоцереброзида при болезни Гоше (см. гл. 316) и концентрации галактозы в крови при недостаточности галактокиназы (см. гл. 314). Нарушение ЕBC может приводить к накоплению как А, так и В, а нарушение ecd — к накоплению А, В и С. При гомоцистинурии вследствие недостаточности цистатионинсинтазы накапливается не только гомоцистин — ближайший предшественник заблокированной реакции, но и метионин—отдаленный ее предшественник (см. гл. 306).
Использование альтернативных путей. Если из-за недостаточности Едв нарушено превращение А в В, то А будет не только накапливаться, но и ускоренно превращаться по обычно минорному альтернативному пути в F и G. Прекрасным примером подобной ситуации может служить фенилкетонурия. Отсутствие активности гидроксилазы фенилаланина приводит к резкой гиперпродукции и экскреции фенилпировиноградной, фенилуксусной и фенилмолочной кислот, т. е. соединений, в норме в крови, и моче не определяемых (см. гл. 306). Если продукты альтернативных путей метаболизма, накапливаясь, служат помехой нормальным клеточным процессам, то усиление этих путей приобретает важное физиологическое значение.
Дефицит продукта. Если физиологически активным продуктом гипотетической последовательности реакций служит D, то блокада любого этапа (от А до D) приведет к недостаточному синтезу D. Тироксин в щитовидной железе образуется именно через такую серию реакций, начиная с транспорта йодида в железу с его последующим окислением и органификацией. К зобному кретинизму, обусловленному нарушением синтеза тироксина, приводит несколько ферментных нарушений. Подобно этому, у ряда больных с врожденной гиперплазией надпочечников на почве нарушения гидроксилирования 21-го углеродного атома стероидного ядра нарушается продукция альдостерона, что приводит к потере соли через почки и гипонатриемическим кризам. Недостаточный синтез продукта может сопровождаться чрезмерным образованием предшественников из-за выпадения механизма регуляции по принципу обратной связи, что происходит при острой интермиттирующей порфирии (DЕAB).
Избыток продукта. Как можно видеть на рис. 305-1, конечный продукт ряда реакций — D — регулирует активность ЕAB — первого фермента этого пути биосинтеза. Нарушение регуляции по принципу обратной связи обнаруживается при нескольких врожденных «ошибках», но биохимические основы этого нарушения не совсем понятны. У некоторых больных с первичной подагрой избыточное образование уратов происходит предположительно из-за дефектности первого фермента пути синтеза пуринов и отсутствия реакции этого фермента на обычные ингибиторы — гипоксантин и аденин. Нарушение регуляции по принципу обратной связи при врожденной гиперплазии надпочечников и врожденном зобном кретинизме обусловлено, вероятно, другими биохимическими механизмами. При этих заболеваниях образование или секреция соответственно адренокортикотропного (АКТГ) и тиреотропного (ТТГ) гормонов не подавляется их обычными «серво»-регуляторами — кортизолом и тироксином, что приводит к гиперплазии и функциональным дефектам в обеих железах-мишенях. Нарушение регуляции по принципу обратной связи — это не единственный механизм, обусловливающий избыток продукта. При заболеваниях, характеризующихся избытком фермента, таких как гиперурикемия, вызванная повышением активности фосфорибозилпирофосфатсинтетазы (см. гл. 309), избыток продукта обусловлен в основном ускорением превращения предшественника в продукт.
Дата добавления: 2015-07-22; просмотров: 874;